
Joint thermal treatment of heavy oil and liquid products of fast wood pyrolysis is investigated. Thermal analysis shows that the coke yield does not increase if the liquid products are added up to 20 mass%. The liquid wood-pyrolysis products decompose much earlier than heavy oil. However, the decomposition of the blends is essentially the same as pure-oil decomposition.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Oil and its refined products are currently the main feedstocks for producing fuels and chemicals. However, it seems interesting to incorporate nontraditional hydrocarbon sources, alternative resources, and renewable feedstocks into petrochemical production to diversify the feedstock base. Furthermore, use of renewable resources in energy is a promising direction.
Heavy oil, reserves of which are already significantly greater than those of traditional oil, is a potential resource for producing petrochemicals and energy. The amounts in Russia alone reach several billion tons [1, 2]. Heavy oils feature high contents of asphaltenes, resins, and other high-molecular-mass compounds with high S and O contents that are responsible for its high viscosity. Therefore, the transportation and refining of heavy oil is a complicated problem [3, 4].
On the other hand, renewable biomass resources can also be used for producing petrochemicals. Fast pyrolysis is an economically effective technology for processing them into feedstock for petrochemical production [5] that uses thermochemical liquefaction of the biomass to produce pyrolysis liquids (PL).
PL are a complicated mixture of decomposition products of cellulose, hemicellulose, and lignin. PL contain organic acids, phenols, aldehydes, furans, carbohydrates, and other compounds [6]. The disadvantages of using PL in energy and refining are low solubility in hydrocarbons, high H2O contents (15-30 wt.%), acidity, high O content (35-40 wt.%), phase separation, and instability [6, 7]. Furthermore, fast PL contain both low-molecular-mass products (up to 30 wt. %) and oligomers of molecular mass up to 5,500 Da with an average molecular mass of 600-1,000 Da [7]. The low-molecular-mass compounds are aldehydes, alcohols, ketones, furans, substituted phenols, carbohydrates, lactones, etc. They are exceedingly reactive even at temperatures below 100°C [8]. As a result, oligomers polymerize and increase their molecular mass and coke and additional amounts of H2O form during distillation at atmospheric pressure or under vacuum.
PL have low viscosities and are polar. As a result, they do not mix with traditional petroleum products [9]. However, joint processing of PL emulsified with heavy oil, which also contains polar compounds, could increase the process efficiency.
Earlier, several proposed possible schemes for refining PL included joint processing with petroleum residuals, in particular, methods based on hydroforming [10,11,12,13,14] and catalytic [15,16,17,18] and thermal cracking [19, 20].
Hydroforming methods remove O via hydrodeoxygenation and hydrocracking of oligomers. Coking also remains problematical because the low-molecular-mass O-containing compounds are exceedingly reactive [8, 10,11,12]. Therefore, classical sulfided catalysts and schemes designed for oil products are unsuitable [21].
Catalytic and thermal cracking methods presuppose reduction of the O content by removing it as CO, CO2, and H2O and reduced molecular masses for the cracking products [16, 17]. Tube furnaces and contact heating apparatuses were used earlier in wood chemistry to distill (crack) wood resin [22, 23] and produce up to 50% oils, fractions of which were used successfully as antioxidants to stabilize cracking-gasolines [24].
Catalytic cracking of PL and oil feedstock was investigated in 2012-2015 at NREL and a pilot plant of FCC PetroBras in Brasilia (200 kg/h) [17, 18] using ENSYN liquid products. The tests lasted over 400 h and produced greater than 3,700 L of catalytic-cracking products, from which diesel fuel and gasoline (1,500 L each) were subsequently produced. The results showed that bio-oil up to 20 wet. % and catalytic-cracking feedstock could be jointly refined in existing FCC systems [17].
Thermal cracking of PL produced from birch wood and heavy oil from a PAO Tatneft field were investigated to study the mechanisms of joint thermal treatment of heavy oil and fast lignocellulose biomass PL and their mutual effects.
Addition of PL at certain concentrations could increase the thermal stability of the mixture during refining. The oil yields could increase through synergism and mutual inhibition of thermal polymerization of heavy-oil compounds and PL. PL, which contain substituted phenols and their oligomers, could inhibit polymerization. Previously, thermal cracking of wood resin on industrial scales produced a wood-resin inhibitor that was highly effective for stabilizing cracking-gasolines and as a thermal polymerization inhibitor [24]. We supposed that PL also contained products that could perform an analogous function.
Heavy oil from a PAO Tatneft field in the Republic of Tatarstan was used in the work. Its principal physicochemical properties are given below:
Dynamic viscosity, mPa·s ..................................................................2480 |
Density, g/cm3H ...............................................................0.9617 |
API ...............................................................15.2 |
Composition of heavy oil SARA-fractions at 293.15 K, wt.s%: |
Volatile constituents ............................................................. 4.2 |
Saturated hydrocarbons ....................................................... 46.1 |
Aromatic hydrocarbons ...................................................... 32.12 |
Resins ....... ........................................................ 12.3 |
Asphaltenes ..................................................................5.3 |
The data indicated that resin and asphaltene fractions that were responsible for the high viscosity made up a significant part of this oil.
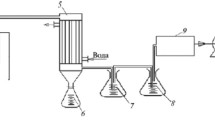
PL obtained from dry birch wood on a UBP-50 pilot unit were used in the work [25]. PL were prepared from ground birch chips at 500 ± 50°C. The moisture content of the dried wood was 8 ± 0.5%. The wood ash content was 0.3 wt.%. The feedstock particle size was 0.5-5 mm with an average size of 1.2 mm. Figure 1 shows a diagram of the unit.

UBP-50 unit: 1) dryer, 2) furnace, 3) pyrolysis chamber, 4) hermetic container, 5) coal bunker, 6) hydraulic valve, 7) flare, 8) blower, 9) condenser, 10) cyclone, 11) heat exchanger, 12) feedstock bunker, 13) fuel tank.
The PL yield was 49% of the mass of dry starting feedstock. Table 1 presents the main properties of the PL.
The specifications of the PL satisfied requirements of ASTM standard D7544-10. The PL were stored in polyethylene containers at 3-5°C.
Thermal analysis of PL, heavy oil, and their mixtures used a TG 209 F1 Libra thermogravimetric analyzer (Netzsch, Germany). Measurements were taken in the range 30-800°C in a dynamic atmosphere of N2 and air (flow rate 75 mL/min) at ramp rate 10 °C/min using open corundum crucibles. The analyzer furnace was calibrated at the melting points of high-purity metals (99.999%) at the flow and ramp rates used in the experiments. Experimental sample masses were 4.5-5 mg. The experiments were described in detail before [26].
Thermal cracking used a laboratory apparatus consisting of a hermetic crucible connected to a condenser for collecting liquids and a gas collector. The crucible was placed into a muffle furnace. The condenser was cooled to ~20°C using tap water.
Thermal cracking at 540°C with a delay time of 15 min was carried out as follows. A crucible with liquid (~1 g) was placed into the muffle furnace and connected hermetically to a condenser and receiving flask. The furnace was heated at the given rate (36°C/min) to 540°C. This rate avoided violent release of volatile compounds and loss of fine feedstock particles from the crucible. The temperature was held at 540°C for 10 min. Then, the crucible was carefully removed from the furnace and cooled to room temperature. The masses of the crucible with the coke residue and the flask with the liquid were measured.
The moisture content of the liquid products was measured during the studies of the thermal-cracking products. The moisture content of PL was measured using Karl Fisher coulometric titration according to GOST 24614-81 on a V20 Compact Volumetric KF Titrator to an accuracy of ±0.06%. The titrant was Karl Fisher Akva M®-Solvent Rapid dissolved in HYDRANAL® -5K. A 10-mL gas chromatography syringe was used for accurate dispensing of wood fast PL. The sample was diluted beforehand in chemically pure i-PrOH in a 1:9 ratio. Then, the sample moisture content was recalculated.
Thermal decomposition of PL, oil, and their mixtures with various PL contents (1, 5, 10, and 20%) was studied in air and N2.
Figure 2 shows that PL underwent thermal decomposition and oxidation significantly earlier than heavy oil. PL decomposition in N2 was practically finished at 350-400°C whereas oil decomposition continued almost to 500°C. However, the fraction of solid residue reached 20 mass% for PL and <10 wt.% for oil. It was concluded that PL decomposed faster and formed a larger amount of solid residue. Both samples combusted completely in air, as confirmed by the 100% mass loss. PL burned completely at lower temperatures than heavy oil.

Thermal analysis in N2 (a) and air (b); mass curves (solid lines) and rates of mass change (dashed lines) of heavy oil (1) and PL (2).
Figure 3 shows mass-loss curves for thermal decomposition and oxidation of PL mixtures and heavy oil in N2 and air. An analysis of the results found that the curves of the mixtures were practically analogous to those of heavy oil even with a PL content of 20% despite the fact that starting PL differed substantially from heavy oil during thermal decomposition and oxidation. The solid residue after thermal treatment was 7-8 wt.% for both heavy oil and a mixture of heavy-oil (80 wt.%) and PL (20 wt.%). This could confirm that addition of PL inhibited thermal polymerization of heavy oil.

Thermal analysis of a mixture of heavy oil (90%) and PL (10%); mass curves (solid lines) and rates of mass change (dashed lines) in N2 (1) and air (2).
The results also suggested that thermal decomposition of the mixtures and heavy oil itself was complete at about the same temperature (485°C).
Thermal decomposition was studied on an experimental unit in addition to the thermogravimetric studies.
Material balance data for thermal cracking of PL and oil were obtained. Figure 4 presents the results.

[labeled Fig. 6 on R. p. 9] Material balance for thermal cracking of PL and oil: 1) liquid products, 2) gases, 3) solid residue, 4) solid residue (from TGA data), 5) H2O.
The results led to the conclusion that adding PL did not increase substantially the coke residue.
The water contents in the liquid products from thermal cracking of PL, oil, and their mixtures were studied. Figure 4 also presents these results.
The coke yields obtained on the laboratory unit were similar to those from thermogravimetric analysis (Fig. 5) and confirmed that the data obtained from the two independent methods were reliable. It can be seen that adding up to 20 wt.% PL to heavy oil did not affect the amount of coke residue.

Coke yield from thermal analysis (1) and data obtained on the laboratory unit (2).
Thermal analysis showed that PL underwent thermal decomposition significantly earlier than heavy oil. However, the decomposition of their mixtures was essentially the same as that of the pure oil. The amounts of coke residue for heavy oil and the mixtures were the same. Oxidation of heavy oil and the mixtures occurred through the same mechanism with the same temperature ranges. All this confirmed that adding PL to heavy oil did not degrade its properties and somewhat inhibited thermal polymerization during joint thermal treatment.
This effect could increase the yield of oil and its stability during thermal cracking of heavy oil feedstocks and biomass PL.
References
L. C. Ortega et al., Energy Fuels, 30, No. 2, 854-863 (2016).
G. P. Kayukova et al., Pet. Chem., 57, No. 8, 657-665 (2017).
T. N. Yusupova et al., Pet. Chem., 57, No. 3, 198-202 (2017).
N. N. Petrukhina et al., Chem. Technol. Fuels Oils, 50, No. 4, 315-326 (2014).
D. Meier et al., Renewable Sustainable Energy Rev., 20, 619-641 (2013).
A. Oasmaa and C. Peacocke, Properties and fuel use of biomass-derived fast pyrolysis liquids. A guide, Espoo, 2010, VTT Publications 731.
S. Zabelkin et al., Constr. Build. Mater., 102, 59-64 (2016).
A. Ardiyanti et al., Appl. Catal., B, 117–118, 105-117 (2012).
M. Ikura et al., Biomass Bioenergy, 24, 221-232 (2003).
Q. Bu et al., Bioresour. Technol., 124, 470-477 (2012).
Elkasabi et al., Fuel Process. Technol., 123, 11-18 (2014).
Rover et al., Fuel, 153, 224-230 (2015).
Wang et al., ACS Catal., 3, No. 5, 1047-1070 (2013).
Huynh et al., J. Sustainable Bioenergy Syst., 5, 151-160 (2015).
Bertero et al., Waste Biomass Valor, 2016; DOI 10.1007/s12649–016–9624-z.
J. F. Garcia et al., “Catalytic cracking of bio-oils improved by the formation of mesopores by means of Y zeolite desilication,” Appl. Catal., A, 503, 1-8 (2015).
Pinho et al., Fuel Process. Technol., 131, 159-166 (2015).
Pinho et al., Fuel, 188, 462-473 (2017).
US Pat. 0152334.
US Pat. 0232164.
V. A. Yakovlev et al., Katal. Prom-sti., No. 4, 48-66 (2012).
V. P. Sumarokov, Chemistry and Technology of Wood Resin Processing [in Russian], Goslesbumizdat, Moscow-Leningrad, 1953, 236 pp.
L. V. Gordon et al., Technology and Equipment of Wood Chemistry Industry: Textbook for Technical Schools [in Russian], 5th Ed., revised, Lesn. Prom-st., Moscow, 1988, 360 pp.
A. N. Zav’yalov et al., Wood Resins and Other Chain Reaction Inhibitors [in Russian], Moscow, 1978, 33 pp.
A. N. Grachev, V. N. Bashkirov, et al., RU Pat. No. 2,395,559, Jul. 27, 2010.
M. A. Varfolomeev et al., Khim. Tekhnol. Topl. Masel, No. 1, 83-88 (2015).
Acknowledgments
The work was supported by a subsidy for state support of Kazan (Volga Region) Federal University to increase its competitiveness among leading global scientific and educational centers.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya i Tekhnologiya Topliv i Masel, No. 5, pp. 6-10, September-October, 2017.
Rights and permissions
About this article

Cite this article
Grachev, A.N., Varfolomeev, M.A., Emel’yanov, D.A. et al. Joint Thermal Treatment of Heavy Oil and Liquid Products of Fast Wood Pyrolysis for Producing Fuels and Chemicals. Chem Technol Fuels Oils 53, 638–645 (2017). https://doi.org/10.1007/s10553-017-0845-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10553-017-0845-z