
Abstract
Purpose
Positive energy imbalance and growth factors linked to obesity promote the phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin (mTOR) pathway. As the obesity–breast cancer associations differ between European American (EA) and African-American (AA) women, we investigated genetic variants in the mTOR pathway and breast cancer risk in these two racial groups.
Methods
We examined 400 single-nucleotide polymorphisms (SNPs) in 31 mTOR pathway genes in the Women’s Circle of Health Study with 1263 incident breast cancers (645 EA, 618 AA) and 1382 controls (641 EA, 741 AA). Multivariable logistic regression was performed separately within racial groups. Effect modification was assessed for measured body size and weight gain since age 20.
Results
In EA women, variants in FRAP1 rs12125777 (intron), PRR5L rs3740958 (synonymous coding), and CDKAL1 rs9368197 (intron) were associated with increased breast cancer risk, while variants in RPTOR rs9900506 (intron) were associated with decreased risk (nominal p-trend for functional and FRAP1 SNPs or p adjusted for correlated test [p ACT] < 0.05). For AA women, variants in RPTOR rs3817293 (intron), PIK3R1 rs7713645 (intron), and CDKAL1 rs9368197 were associated with decreased breast cancer risk. The significance for FRAP1 rs12125777 and RPTOR rs9900506 in EA women did not hold after correction for multiple comparisons. The risk associated with FRAP1 rs12125777 was higher among EAs who had body mass index ≥30 kg/m2 (odds ratio = 7.69, 95 % CI 2.11–28.0; p-interaction = 0.007) and gained weight ≥35 lb since age 20 (odds ratio = 3.34, 95 % CI 1.42–7.85; p-interaction = 0.021), compared to their counterparts.
Conclusions
The mTOR pathway may be involved in breast cancer carcinogenesis differently for EA and AA women.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Obesity has been implicated in the development of breast cancer [1]. Body mass index (BMI), a measurement of general obesity, is positively associated with breast cancer risk among postmenopausal women, but not in premenopausal women [1, 2]. The risk in postmenopausal women may also vary by breast cancer subtype defined by hormone receptor status. BMI has been more often associated with an increased risk of estrogen receptor positive (ER+) than of ER negative (ER−) breast cancer in postmenopausal women [2]. These observations are mainly derived from women of European ancestry. Among postmenopausal African-American (AA) women, BMI is positively associated with ER+ breast cancer risk. However, it is inversely associated with ER− and triple-negative (ER−, progesterone receptor [PR] negative, and human epidermal growth factor receptor [Her-2/neu] negative) breast cancer risks in postmenopausal AA women [3, 4]. In addition, waist-to-hip ratio (WHR), an indicator of abdominal obesity, may be important in increasing risk of both ER subtypes of breast cancer [4] in both pre- and postmenopausal AA women [4–7]. Although several hypotheses that incorporate hormonal and inflammatory factors have been proposed, the mechanisms underlying the influence of obesity on breast cancer risk are not fully understood [8].
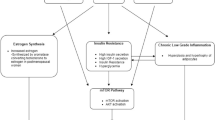
Among various obesity-related pathological pathways, the phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin (PI3K-AKT-mTOR or mTOR) pathway has been linked to cancer hallmarks [9]. Because a key role of the mTOR pathway is to maintain cellular energy homoeostasis, it is activated when the energy content of the diet is greater than energy expended by the body, i.e., positive energy imbalance [10]. The regulation of energy homeostasis occurs in the mTOR complex 1 (mTORC1), which initiates various downstream responses, including modulation of ER transcriptional activity (Fig. 1) [9, 11]. In addition, growth factors, e.g., glucose and insulin, can signal mTOR complex 2 (mTORC2) and influence cell metabolism and survival through AKT (protein kinase B). In breast cancer, somatic mutations in the mTOR pathway are commonly observed [12]. A few studies have examined associations between genetic polymorphisms in the mTOR pathway and breast cancer risk among European women [13, 14]. However, these studies have focused only on a small number of genes (i.e., TSC1, TSC2, and LAMTOR). It is currently unknown whether the genetic variants in other key mTOR pathway elements, such as the mTOR coding gene FRAP1, affect breast cancer risk. In addition, the role of obesity in the possible association of the mTOR pathway with breast cancer risk warrants investigation, especially since a positive energy imbalance has been shown to exert effect modification on the association between mTOR pathway genes and risk of kidney and bladder cancers [15, 16].

Overview of the mTOR pathway. 4E-BP1 4E-binding protein-1, eIF-4E eukaryotic initiation factor-4E, EGFR epidermal growth factor receptor, ER estrogen receptor, HER2 receptor tyrosine-protein kinase erbB-2, IGF-R1 insulin-like growth factor 1 receptor, IRS insulin receptor substrate, PRAS40 proline-rich Akt substrate 40 kDa, PTEN phosphatase and tensin homolog, S6 40S ribosomal protein, S6K1 S6 kinase 1
The objective of this study was to assess the association of genetic variants in the mTOR pathway with breast cancer risk. Given that the mTOR pathway can be activated by positive energy imbalance and growth factors, both of which are directly associated with obesity and weight gain, we examined whether BMI, WHR, and weight gain in adulthood modified the association of genetic variants in the mTOR pathway and breast cancer risk. Because there are differences in breast cancer risks associated with obesity between race, ER status, and menopausal status, we analyzed the data for European American (EA) and AA women separately and conducted exploratory stratified analyses according to ER and menopausal status.
Methods
Study population
The Women’s Circle of Health Study (WCHS) is an ongoing multisite case–control study in New York City and New Jersey. WCHS was designed to evaluate genetic and lifestyle risk factors for both early and aggressive breast cancer and to compare the distribution of these factors in EA and AA women [17, 18]. Breast cancer cases were women with primary, histologically confirmed invasive breast cancer or ductal carcinoma in situ (DCIS) diagnosed within 9 months of recruitment. Other inclusion criteria were self-identified EA and AA women, 20–75 years of age, literate in English, and with no previous history of cancer other than non-melanoma skin cancer. In New York City, cases were identified through collaborations with hospitals that had a large AA patient base, and in NJ, cases were ascertained through the State Cancer Registry using rapid case ascertainment. Controls had the same inclusion criteria plus no history of cancer and were identified during the same time period as the cases. Controls were ascertained by random digit dialing and frequency matched with cases by 5-year age groups, race, and telephone exchanges (area code plus 3-digit prefixes, for cases from New York City) or county of residence (for cases from New Jersey). In New Jersey, AA controls were also recruited through outreach sources such as health events [18]. Information on ER status was from pathology reports. Questionnaire, anthropometric, and genetic data from 1279 cases (658 EA, 621 AA) and 1392 controls (649 EA, 743 AA) recruited during 2002–2011 were available from WCHS. Of these, 26 women (16 cases and 10 controls) were excluded due to missing data on BMI or other covariates, leaving 1,263 cases (645 EA, 618 AA) and 1,382 controls (641 EA, 741 AA) in the statistical analyses of genetic associations. In addition, 68 women (41 cases and 27 controls) with missing data on WHR and weight gain were excluded, leaving 1,222 cases (634 EA, 588 AA) and 1,355 controls (631 EA, 724 AA) women in the effect modification analysis. The WCHS protocol was approved by institutional review boards at Roswell Park Cancer Institute, the Cancer Institute of New Jersey, Mount Sinai School of Medicine, and participating hospitals in New York. Signed informed consent was obtained from each participant prior to interview and biospecimen collection.
DNA collection and genotyping
Blood samples were initially collected for DNA extraction, but after enrollment of approximately 850 participants, saliva samples were collected as a source of DNA due to cost restrains. Genomic DNA was extracted from whole blood using the FlexiGeneTM DNA isolation kits (Qiagen Inc., Valencia, CA) and, from saliva, Oragene™ kits (DNA Genotek Inc., Kanata, Ontario, Canada). DNA was evaluated and quantitated by NanoDrop UV spectrometer (Thermo Fisher Scientific Inc., Wilmington, DE) and PicoGreen-based fluorometric assay (Molecular Probes, Invitrogen Inc., Carlsbad, CA), and stored at −80 °C until analysis.
We selected 27 protein-coding genes in the mTOR pathway (Supplementary Table 1) and additionally, four insulin and obesity-related genes (CDKAL1, CYB5R4, GIP, and IRS1) known to have upstream influences on energy-balance regulation [19]. We selected a set of representative single-nucleotide polymorphisms (tagSNPs) that had high correlation (r 2 ≥ 0.8) with other variants within a given linkage disequilibrium (LD) bin, were present at a minor allele frequency (MAF) of ≥0.05 in at least one reference population, and were within 10 kb upstream of the 5′-untranslated region (UTR) and 10 kb downstream of the 3′-UTR of the gene using SNPinfo [20]. Within SNPinfo, we employed reference populations (CEU and ASW) in HapMap2 to determine the LD structure and allelic correlations [21]. In addition, we also included SNPs with either known or predicted functional changes based on the literature. A total of 425 SNPs were attempted for genotyping.
Genotyping was performed using the Illumina GoldenGate platform (Illumina Inc., San Diego, CA) at Roswell Park Cancer Institute. For quality control, 5 % blinded duplicates and two sets of in-house trio samples were included across all plates. The concordance among blind duplicate pairs was greater than 99.9 %. SNPs were excluded if they had <90 % call rate, <5 % MAF for both EAs and AAs, or Hardy–Weinberg equilibrium (HWE) p < 0.0001. Subsequently, 400 SNPs passed these filters and were included in statistical analyses (Supplementary Table 6). LD structures of the samples were assessed among EA and AA controls separately using Genome Variation Server 138 (University of Washington, Seattle, WA). To account for population admixture, all samples were also genotyped for a validated panel of 100 ancestry informative markers (AIMs) [22]. Based on the AIM data, proportions of European ancestry and African ancestry for each individual were computed using the Bayesian Markov chain Monte Carlo clustering algorithm implemented in the STRUCTURE program [23].
Epidemiological data collection
In-person interviews and anthropometric measurements were conducted with a standardized protocol [3]. The interview covered a comprehensive set of established and suspected risk factors for breast cancer. For the current study, data on demographics, history of benign breast disease, reproductive and menstrual histories, family history of breast cancer in a first-degree relative and cigarette smoking were utilized. Interviewers also queried participants’ weight and height 1 year before diagnosis (for cases) or reference date (for controls) and at every 10 years since the age of 20. Anthropometric measurements were taken at the end of the interview by trained staff with standing height measured once to the nearest 0.1 cm. To minimize measurement error, waist and hip circumferences were measured twice to the nearest 0.1 cm; a third measurement was taken if the difference between the first and second measurement was >2 cm. The two (or three) measurements were averaged for analyses. Measurements were not taken due to refusal, pregnancy >3 months, or physical impairments.
Statistical analyses
Characteristics of cases and controls were compared with t tests for continuous variables and Chi-square tests for categorical variables. Logistic regression was used to estimate odds ratios (OR) and 95 % confidence intervals (CI) adjusted for age, proportion of European ancestry, education, BMI, family history of breast cancer, history of benign breast disease, age at menarche, parity, breastfeeding, menopausal status, and smoking status. We adjusted for the proportion of European ancestry only in models for both EA and AA women. We did not adjust the proportion of African ancestry for AA women because the proportions of African ancestry and European ancestry add up to 1 in our data, and thus, the results would have remained the same if adjusting for African ancestry. A more general codominant inheritance model was initially assumed to avoid imposing a structure on the response expected for each additional copy of the variant allele. We also tested a dominant model, but did not test recessive models because the number of participants homozygous for the rare variant allele was small for some SNPs based on genotype distributions in EA women. p values for trend were calculated by coding variant alleles based on the number of rare alleles (0, 1, 2) and analyzed as a continuous variable in regression to subsequently test for an additive model. p values for trend were adjusted for correlated tests (p ACT) at the gene-level to account for multiple comparisons [24]. We prioritized all 15 SNPs (two non-synonymous) in FRAP1, our main hypothesized gene, plus 14 non-synonymous SNPs and 35 high-functional SNPs, i.e., RegulomeDB score from 1 to 3 (Supplementary Table 7) [25], in the other genes. A nominal p-trend <0.05 was considered significant for this set of SNPs. For tagSNPs, a p ACT < 0.05 was considered significant. For significant SNPs, we further estimated ORs for ER+ and ER− breast cancer risk. p values for heterogeneity were calculated using case–case logistic regression models comparing ER subtypes. Because the MAFs and LD structure in a large proportion of SNPs differed between EAs and AAs (Supplementary Table 6), all analyses were conducted separately by race.
To assess effect modification on genetic associations by obesity, we conducted stratified analyses by BMI, WHR, and weight change since the age of 20 years for the significant SNPs. BMI was calculated as the measured weight (kg) divided by height (m)2 and categorized as <25 (underweight to normal), 25–<30 (overweight), and ≥30 kg/m2 (obese), according to the World Health Organization (WHO) International Classification. WHR was also calculated from the measured values of waist and hip circumferences and categorized into ≤0.85 (normal) and >0.85 (abdominal obesity) [26]. Weight change since age 20 was derived from current measured weight minus self-reported weight at age 20. Weight change was a priori categorized into <10, 10–<25, 25–<35, ≥35 lb. BMI, WHR, and adulthood weight gain were mutually adjusted to reduce the influence of the other two variables on risk estimates. A sensitivity analysis without the mutual adjustment of body size and weight gain was performed because these measurements may be collinear. p values for interaction were obtained using Wald tests for the product term of SNP and body size or weight gain variables; 36 tests were performed (six SNPs times three body size/weight gain variables times two racial groups). A combined EA and AA model was built to assess the interaction between race and SNPs. Statistical analyses were performed using SAS 9.4 software. Functional exploration of significant SNPs was performed using the Encyclopedia of DNA Elements (ENCODE) data [27].
Results
Table 1 presents the characteristics of cases and controls separately for EA and AA women. EA cases were more likely to have a first-degree relative diagnosed with breast cancer (p = 0.001), but less likely to have a college or postgraduate degree (p < 0.001) and to breastfeed (p = 0.008), compared to EA controls. Among both cases and controls, AAs were more likely to be obese than EAs (52.1 vs. 28.0 % in cases, and 53.2 vs. 27.0 % in controls). For both EAs and AAs, cases were more likely to have history of benign breast disease (p = 0.001 and p < 0.001, respectively). For AA women, there were a higher proportion of current smokers in controls than cases (p < 0.001). The categories of BMI, WHR, and weight gain from age 20 did not differ by case status in either racial group. Among those with known ER status of breast tumors, AA cases had a higher proportion of ER− tumors compared to EA cases (31.5 vs. 17.2 %).
Table 2 lists associations of SNPs with breast cancer in either EA or AA women. In FRAP1, the variant A allele of rs12125777 was associated with a higher risk of breast cancer in EAs (ORGA/AA vs. GG = 1.69, 95 % CI 1.08–2.64; nominal p-trend = 0.020; p ACT = 0.17), but not in AAs. In regulatory associated protein of mTOR complex 1 (RPTOR), two SNPs that potentially have regulatory functions were associated with breast cancer risk. The variant G allele of RPTOR rs9900506 (RegulomeDB score = 3a) was associated with a lower breast cancer risk in EA women (ORGG vs. AA = 0.68, 95 % CI 0.48–0.95; nominal p-trend = 0.042; p ACT = 0.89), but not in AA women. However, the variant of RPTOR rs3817293 (RegulomeDB score = 2b) was associated with a lower breast cancer risk in AA women (ORAA/GA vs. GG = 0.79, 95 % CI 0.63–0.99; nominal p-trend = 0.044; p ACT = 0.97), but not in EA women. Among the other tagSNPs, the variant allele of rs7713645 in the phosphoinositide-3-kinase, regulatory subunit 1 (PIK3R1) was associated with a lower risk of breast cancer in AA women (ORCC vs. AA = 0.56, 95 % CI 0.33–0.97; p ACT = 0.041), while proline-rich 5-like (PRR5L) rs3740958 was associated with a higher risk in EA women (ORGG/GA vs. AA = 1.71, 95 % CI 1.29–2.27 for; p ACT = 0.005). In addition, CDK5 regulatory subunit-associated protein 1-like 1 (CDKAL1) rs9368197 was associated with a higher risk of breast cancer in EA women (ORAA vs. CC = 1.66, 95 % CI 1.08–2.54; nominal p-trend 0.032; p ACT was not applicable because only one SNP was genotyped in this gene). The increased risk was mainly driven by the increase in risk of ER + tumors, and not of ER– tumors (p-heterogeneity = 0.021; Supplementary Table 2). However, the association was reversed in AA women (ORAA/AC vs. CC = 0.70, 95 % CI 0.51–0.97; nominal p-trend = 0.015). The associations with overall breast cancer were significantly different between EA and AA women for PRR5L rs3740958 and CDKAL1 rs9368197 (p-interaction of SNP and race <0.001 and 0.005, respectively).
Among these SNPs associated with breast cancer risk, significant effect modification of BMI and weight gain was observed for FRAP1 rs12125777 in EA women (Table 3). Our data showed that among EA women, the association of GA/AA vs. GG genotypes and breast cancer risk was stronger among overweight women (25 ≤ BMI < 30; OR 2.37, 95 % CI 0.91–6.13) and obese women (BMI ≥ 30; OR 7.69, 95 % CI 2.11–28.0), compared to normal/underweight women (BMI < 25; OR 0.85, 95 % CI 0.45–1.61; p-interaction = 0.007). The 95 % CIs, however, were wide due to a small number of cases in women with the rare allele. A similar pattern of effect modification was also observed for weight gain since age 20. The association for those carrying the variant A allele of rs12125777 was strengthened among EA women who gained 35 lbs. or more since age 20 (OR 3.34, 95 % CI 1.42–7.85), compared to the associations among those with lesser weight gain (p-interaction = 0.021). In the model without mutual adjustment of the three variables, the risk estimates were more conservative and 95 % confidence intervals were narrower (e.g., OR 5.68, 95 % CI 1.78–18.1 for GA/AA vs. GG in BMI ≥ 30 among EA women), compared to models with mutual adjustment (OR 7.69, 95 % CI 2.11–28.0) (Supplementary Table 3). The observed effect modification of body size and weight gain seemed more pronounced for ER− breast cancer risk than for ER+ breast cancer risk (Supplementary Table 4), although the sample size was small after the stratification, and thus, these estimates were likely not precise. Effect modification of body size and weight gain was not observed in AA women for this SNP (Table 3), or for the other significant SNPs in either racial group (data not shown).
Discussion
In this study, FRAP1 rs12125777, RPTOR rs9900506 and rs3817293, PIK3R1 rs7713645, PRR5L rs3740958, and CDKAL1 rs9368197 were associated with breast cancer risk in either EA or AA women. The associations for FRAP1 and RPTOR SNPs in EA women were not significant after adjusting for multiple comparisons and thus should be interpreted with caution. With a limited number of tests on the hypothesized gene–environment interaction, we observed a potential effect modification of BMI and weight gain on the association of FRAP1 rs12125777 with breast cancer risk in EA women. To our knowledge, this is the first study to examine the relationship between the mTOR pathway and breast cancer risk in both EA and AA women and to assess potential gene–environment interactions.
mTOR and RPTOR are core protein elements of mTORC1 and our data suggest that three SNPs in their coding genes may be associated with breast cancer risk. Based on the ENCODE data, FRAP1 rs12125777 is located in a region with active transcriptional promoters in mammary epithelial cells and DNase hypersensitivity sites in mammary gland (Supplementary Table 5). The SNP also maps a region potentially altering the motifs of ER-alpha expression. Two correlated SNPs, FRAP1 rs2536 (3′-UTR) and rs12116957 (intron), also map to regions annotated with many transcription factors. RPTOR rs9900506 is located in a region with weak transcriptional enhancers. RPTOR rs9900506 was examined for the risk of renal cell carcinoma, although there was no evidence of an association [15]. The regulatory function of RPTOR rs3817293 is unclear. These SNPs in FRAP1 and RPTOR have not been reported in studies of breast cancer risk and warrant replication.
We also observed that the variant of rs3740958 in PRR5L, also known as PROTOR2, was associated with increased breast cancer risk in EA women. PROTOR2 protein is part of mTORC2. PRR5L rs3740958 is a synonymous coding SNP and located within known functional elements. According to ENCODE data, rs3740958 overlaps transcription regulatory (both promoter and enhancer) marks in many cell types, although it is unclear whether they exist in mammary epithelial and myoepithelial cells. PRR5L rs3740958 is not in high LD with other SNPs in either the populations from the 1000 Genomes Project or in our study population, and has not been reported in studies in relation to the risk of cancer or other outcomes.
PIK3R1 encodes an 85 kD regulatory subunit of PI3K. PI3K plays an important role in the metabolic actions of insulin, and variants in this gene have been associated with insulin resistance and type 2 diabetes [28, 29]. The variant allele of PIK3R1 rs7713645 has been linked to a higher BMI and elevated glucose levels in European women [30]. In our study, however, the SNP variant was associated with a decreased risk of breast cancer in AA women. It is noteworthy that the risk associated with this SNP may differ by the ER status of tumors. The decreased risk was mainly driven by ER+ tumors (OR 0.59, 95 % CI 0.32–1.09). However, for ER− breast cancer, the SNP was associated with a non-significant increased risk (OR 2.14, 95 % CI 0.62–7.35; p-heterogeneity = 0.09; Supplementary Table 2). Because the risk allele (C) is the major allele in AA women, it is important to confirm whether the variant is associated with an increased risk of ER– breast cancer.
CDKAL1 rs9368197 was the only SNP associated with breast cancer risk in both EA and AA women. However, the direction of association was heterogeneous across the two populations. CDKAL1 encodes a subunit of cyclin-dependent protein kinase 5 (CDK5), involving posttranscriptional RNA modification [31]. CDK5 is overexpressed in breast cancer [32]. Several variants in CDKAL1 have been found to be associated with type 2 diabetes and obesity [33, 34]. CDKAL1 rs9368197 potentially confers adaptations in energy-balance regulation in East Asians [35]. As we cannot eliminate the possibility that our findings were due to chance, whether the SNP in relation to breast cancer etiology differs between racial groups requires further research.
We observed that the association of FRAP1 rs12125777 with increased breast cancer risk was stronger in EA women with overweight or obese and greater weight gain than those with normal BMI and less weight gain. These findings provide some evidence for our hypothesis that the mTOR pathway, which is promoted by positive energy imbalance and increased growth factors associated with obesity, plays a role in the development of breast cancer. In animal models, an over-activated mTOR pathway due to obesity accelerates breast tumor growth [36]. A necessary next step in exploring the role of FRAP1 rs12125777 is to confirm whether the SNP or its mapped region has any functional impact on the mTOR pathway among EA women. Because among EA women, the positive association between obesity and breast cancer risk was more prominent for ER+ and postmenopausal breast cancer than ER− and premenopausal breast cancer [2], we explored effect modification of body size and weight gain according to ER subtype and menopausal status. In this exploratory analysis, the effect modification on FRAP1 rs12125777 remained significant for ER+ breast cancer, although the association seemed to be weaker than for ER− (Supplementary Table 4). Also, the SNP–breast cancer association appeared to be larger in postmenopausal women with BMI ≥ 30 kg/m2 (for all breast cancer ORGA/AA vs. GG = 12.5, 95 % CI 1.42–110) than premenopausal obese women (ORGA/AA vs. GG = 5.05, 95 % CI 0.94–27.2; data not shown). These risk estimates are likely inflated by chance due to small number of cases in the exposure group (the GA and AA genotypes) after stratification, but the data provide some hints that the mTOR pathway may be important for ER+ and postmenopausal breast cancer risk in EA women. A larger study is warranted to confirm these observations. For AA women, however, it was less clear whether the SNP can be influenced by body size and weight gain. Because our investigation on effect modification was restricted to a small number of SNPs with significant main effects, we may have missed risk SNPs or loci that would interact with body size among AA women.
Different associations of genetic variants with breast cancer between EA and AA in terms of strength or direction are commonly observed [37, 38]. In the current study, only one SNP (CDKAL1 rs9368197) was significantly associated with breast cancer risk in both EA and AA women and the directions of associations for two SNPs in PRR5L and CDKAL1 were different between the two groups. The underlying reasons of these racial differences in genetic associations are likely multifactorial. As the LD structures are distinct between EA and AA women [39], with the SNP tagging approach, it is possible that the studied SNPs are linked to the causal variants only in either EA or AA women, but not both. Also, the often-observed differences in frequencies of minor alleles between EA and AA women can lead to differences in statistical power, and thus, true associations may not be observed. Our analysis on effect modification by body size also suggests different results across the two racial groups. In addition to the differences in genetic structures between populations, the relationships of body size with body composition are also different between EA and AA women. For example, for a given amount of body fat, which is correlated with BMI [40], EA women tend to have more visceral adipose tissue (and less subcutaneous adipose tissue), than AA women [41]. Visceral adipose tissue has been known to be active in signaling the insulin-like growth factor pathway [42], and potentially, the mTOR pathway [43]. Thus, it is possible that the measurements of body size represent these factors to different extents among EA and AA women and contribute to the heterogeneous effect modification of body size in our study.
In the effect modification analysis, we mutually adjusted for BMI, WHR, and weight gain to simultaneously account for the effects of each of the variables. However, these three variables are likely correlated with each other, and thus, the analysis may be subject to multicollinearity and the 95 % CIs might have been inflated. We examined this issue by estimating the variance inflation factor (VIF) using linear regression; the VIFs were 2.26 for BMI, 1.18 for WHR, and 2.18 for weight gain variables. Thus, the potential inflation of standard errors (square root of VIF) for risk estimates due to the multicollinearity is likely limited, except for BMI. Although the 95 % CIs of the genetic association in the strata of BMI are wider in the mutual adjustment model than in the model without the mutual adjustment, both models show significant effect modification.
Our study has several strengths. This study is a more comprehensive evaluation of mTOR pathway SNPs in relation to breast cancer risk than most previous studies [13, 14]. The approximately equal number of EA and AA women in the WCHS allows for investigations of etiological factors of breast cancer within EA and AA women separately. This is important because the distribution of breast cancer subtypes is different between these two populations and etiological factors may contribute to breast cancer risk differently. Also, WCHS collected in-depth information on covariates and most anthropometric measurements were taken by trained staff and were thus less prone to measurement error than self-report.
Several limitations should be noted. First, SNPs identified as being related to breast cancer risk in our study may not be causal, as the discovery of causal SNPs requires fine mapping efforts. The prioritized or functional SNPs that were not significant after the correction of multiple comparisons warrant further confirmation. Second, the information on ER status relied on pathology reports from different institutions, but any misclassification is unlikely to differ by genotype. Third, case–control studies are subject to selection bias, whereby controls do not adequately represent the source population from which the cases arose. To improve representativeness, we recruited AA controls from communities to supplement those recruited using random digit dialing [18]. In addition, the number of breast cancer cases was small within strata of body size and weight gain, resulting in wide 95 % CIs and potentially inflated risk estimates.
In conclusion, we observed associations with breast cancer risk in either EA or AA women for SNPs in FRAP1, RPTOR, PIK3R1, and PRR5L in the mTOR pathway and an obesity-linked SNP in CDKAL1. We cannot eliminate the possibility that the associations of FRAP1 rs12125777 and RPTOR rs9900506 in EA women might be due to the elevated false positive rate from multiple comparisons. The association of SNP of FRAP1 rs12125777 with breast cancer risk in EA women may be modified by body size and weight gain. Contingent on confirmation and validation, our findings suggest that carcinogenic signaling related to the mTOR pathway may differentially influence breast cancer risk among EA and AA women.
References
World Cancer Research Fund/American Institute for Cancer Research (2010) Continuous update project report. Food, Nutrition, Physical Activity, and the Prevention of Breast Cancer
Munsell MF, Sprague BL, Berry DA, Chisholm G, Trentham-Dietz A (2014) Body mass index and breast cancer risk according to postmenopausal estrogen-progestin use and hormone receptor status. Epidemiol Rev 36(1):114–136. doi:10.1093/epirev/mxt010
Bandera EV, Chandran U, Zirpoli G, Gong Z, McCann SE, Hong CC, Ciupak G, Pawlish K, Ambrosone CB (2013) Body fatness and breast cancer risk in women of African ancestry. BMC Cancer 13:475. doi:10.1186/1471-2407-13-475
Bandera EV, Chandran U, Hong CC, Troester MA, Bethea TN, Adams-Campbell LL, Haiman CA, Park SY, Olshan AF, Ambrosone CB, Palmer JR, Rosenberg L (2015) Obesity, body fat distribution, and risk of breast cancer subtypes in African American women participating in the AMBER Consortium. Breast Cancer Res Treat 150(3):655–666. doi:10.1007/s10549-015-3353-z
White AJ, Nichols HB, Bradshaw PT, Sandler DP (2015) Overall and central adiposity and breast cancer risk in the sister study. Cancer. doi:10.1002/cncr.29552
Harvie M, Hooper L, Howell AH (2003) Central obesity and breast cancer risk: a systematic review. Obes Rev 4(3):157–173
Amadou A, Ferrari P, Muwonge R, Moskal A, Biessy C, Romieu I, Hainaut P (2013) Overweight, obesity and risk of premenopausal breast cancer according to ethnicity: a systematic review and dose-response meta-analysis. Obes Rev 14(8):665–678. doi:10.1111/obr.12028
Vucenik I, Stains JP (2012) Obesity and cancer risk: evidence, mechanisms, and recommendations. Ann N Y Acad Sci 1271:37–43. doi:10.1111/j.1749-6632.2012.06750.x
Zoncu R, Efeyan A, Sabatini DM (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12(1):21–35. doi:10.1038/nrm3025
Hall KD, Heymsfield SB, Kemnitz JW, Klein S, Schoeller DA, Speakman JR (2012) Energy balance and its components: implications for body weight regulation. Am J Clin Nutr 95(4):989–994. doi:10.3945/ajcn.112.036350
Rugo HS, Keck S (2012) Reversing hormone resistance: have we found the golden key? J Clin Oncol 30(22):2707–2709. doi:10.1200/JCO.2012.42.1271
Martin LA, Andre F, Campone M, Bachelot T, Jerusalem G (2013) mTOR inhibitors in advanced breast cancer: ready for prime time? Cancer Treat Rev 39(7):742–752. doi:10.1016/j.ctrv.2013.02.005
De Araujo ME, Erhart G, Buck K, Muller-Holzner E, Hubalek M, Fiegl H, Campa D, Canzian F, Eilber U, Chang-Claude J, Coassin S, Haun M, Kedenko L, Paulweber B, Reitsamer R, Himmel I, Flesch-Janys D, Lamina C, Kronenberg F, Huber LA, Kloss-Brandstatter A (2013) Polymorphisms in the gene regions of the adaptor complex LAMTOR2/LAMTOR3 and their association with breast cancer risk. PLoS ONE 8(1):e53768. doi:10.1371/journal.pone.0053768
Mehta MS, Vazquez A, Kulkarni DA, Kerrigan JE, Atwal G, Metsugi S, Toppmeyer DL, Levine AJ, Hirshfield KM (2011) Polymorphic variants in TSC1 and TSC2 and their association with breast cancer phenotypes. Breast Cancer Res Treat 125(3):861–868. doi:10.1007/s10549-010-1062-1
Shu X, Lin J, Wood CG, Tannir NM, Wu X (2013) Energy balance, polymorphisms in the mTOR pathway, and renal cell carcinoma risk. J Natl Cancer Inst 105(6):424–432. doi:10.1093/jnci/djt005
Lin J, Wang J, Greisinger AJ, Grossman HB, Forman MR, Dinney CP, Hawk ET, Wu X (2010) Energy balance, the PI3K-AKT-mTOR pathway genes, and the risk of bladder cancer. Cancer Prev Res (Phila) 3(4):505–517. doi:10.1158/1940-6207.CAPR-09-0263
Ambrosone CB, Ciupak GL, Bandera EV, Jandorf L, Bovbjerg DH, Zirpoli G, Pawlish K, Godbold J, Furberg H, Fatone A, Valdimarsdottir H, Yao S, Li Y, Hwang H, Davis W, Roberts M, Sucheston L, Demissie K, Amend KL, Tartter P, Reilly J, Pace BW, Rohan T, Sparano J, Raptis G, Castaldi M, Estabrook A, Feldman S, Weltz C, Kemeny M (2009) Conducting molecular epidemiological research in the age of HIPAA: a multi-institutional case–control study of breast cancer in African-American and European-American women. J Oncol 2009:871250. doi:10.1155/2009/871250
Bandera EV, Chandran U, Zirpoli G, McCann SE, Ciupak G, Ambrosone CB (2013) Rethinking sources of representative controls for the conduct of case–control studies in minority populations. BMC Med Res Methodol 13:71. doi:10.1186/1471-2288-13-71
Chang CL, Cai JJ, Cheng PJ, Chueh HY, Hsu SY (2011) Identification of metabolic modifiers that underlie phenotypic variations in energy-balance regulation. Diabetes 60(3):726–734. doi:10.2337/db10-1331
Xu Z, Taylor JA (2009) SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res 37(Web Server issue):W600–W605. doi:10.1093/nar/gkp290
International HapMap Consortium, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, Hu H, Hu W, Li C, Lin W, Liu S, Pan H, Tang X, Wang J, Wang W, Yu J, Zhang B, Zhang Q, Zhao H, Zhou J, Gabriel SB, Barry R, Blumenstiel B, Camargo A, Defelice M, Faggart M, Goyette M, Gupta S, Moore J, Nguyen H, Onofrio RC, Parkin M, Roy J, Stahl E, Winchester E, Ziaugra L, Altshuler D, Shen Y, Yao Z, Huang W, Chu X, He Y, Jin L, Liu Y, Sun W, Wang H, Wang Y, Xiong X, Xu L, Waye MM, Tsui SK, Xue H, Wong JT, Galver LM, Fan JB, Gunderson K, Murray SS, Oliphant AR, Chee MS, Montpetit A, Chagnon F, Ferretti V, Leboeuf M, Olivier JF, Phillips MS, Roumy S, Sallee C, Verner A, Hudson TJ, Kwok PY, Cai D, Koboldt DC, Miller RD, Pawlikowska L, Taillon-Miller P, Xiao M, Tsui LC, Mak W, Song YQ, Tam PK, Nakamura Y, Kawaguchi T, Kitamoto T, Morizono T, Nagashima A, Ohnishi Y, Sekine A, Tanaka T, Tsunoda T, Deloukas P, Bird CP, Delgado M, Dermitzakis ET, Gwilliam R, Hunt S, Morrison J, Powell D, Stranger BE, Whittaker P, Bentley DR, Daly MJ, de Bakker PI, Barrett J, Chretien YR, Maller J, McCarroll S, Patterson N, Pe’er I, Price A, Purcell S, Richter DJ, Sabeti P, Saxena R, Schaffner SF, Sham PC, Varilly P, Stein LD, Krishnan L, Smith AV, Tello-Ruiz MK, Thorisson GA, Chakravarti A, Chen PE, Cutler DJ, Kashuk CS, Lin S, Abecasis GR, Guan W, Li Y, Munro HM, Qin ZS, Thomas DJ, McVean G, Auton A, Bottolo L, Cardin N, Eyheramendy S, Freeman C, Marchini J, Myers S, Spencer C, Stephens M, Donnelly P, Cardon LR, Clarke G, Evans DM, Morris AP, Weir BS, Mullikin JC, Sherry ST, Feolo M, Skol A, Zhang H, Matsuda I, Fukushima Y, Macer DR, Suda E, Rotimi CN, Adebamowo CA, Ajayi I, Aniagwu T, Marshall PA, Nkwodimmah C, Royal CD, Leppert MF, Dixon M, Peiffer A, Qiu R, Kent A, Kato K, Niikawa N, Adewole IF, Knoppers BM, Foster MW, Clayton EW, Watkin J, Muzny D, Nazareth L, Sodergren E, Weinstock GM, Yakub I, Birren BW, Wilson RK, Fulton LL, Rogers J, Burton J, Carter NP, Clee CM, Griffiths M, Jones MC, McLay K, Plumb RW, Ross MT, Sims SK, Willey DL, Chen Z, Han H, Kang L, Godbout M, Wallenburg JC, L’Archeveque P, Bellemare G, Saeki K, An D, Fu H, Li Q, Wang Z, Wang R, Holden AL, Brooks LD, McEwen JE, Guyer MS, Wang VO, Peterson JL, Shi M, Spiegel J, Sung LM, Zacharia LF, Collins FS, Kennedy K, Jamieson R, Stewart J (2007) A second generation human haplotype map of over 3.1 million SNPs. Nature 449 (7164):851–861. doi:10.1038/nature06258
Ruiz-Narvaez EA, Rosenberg L, Wise LA, Reich D, Palmer JR (2011) Validation of a small set of ancestral informative markers for control of population admixture in African Americans. Am J Epidemiol 173(5):587–592. doi:10.1093/aje/kwq401
Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000) Association mapping in structured populations. Am J Hum Genet 67(1):170–181. doi:10.1086/302959
Conneely KN, Boehnke M (2007) So many correlated tests, so little time! Rapid adjustment of P values for multiple correlated tests. Am J Hum Genet 81(6):1158–1168. doi:10.1086/522036
Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, Cherry JM, Snyder M (2012) Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 22(9):1790–1797. doi:10.1101/gr.137323.112
World Health Organization (2011) Waist circumference and waist–hip ratio. Report of a WHO expert consultation, Geneva, 8–11 December 2008
Encode Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489(7414):57–74. doi:10.1038/nature11247
Baynes KC, Beeton CA, Panayotou G, Stein R, Soos M, Hansen T, Simpson H, O’Rahilly S, Shepherd PR, Whitehead JP (2000) Natural variants of human p85 alpha phosphoinositide 3-kinase in severe insulin resistance: a novel variant with impaired insulin-stimulated lipid kinase activity. Diabetologia 43(3):321–331
Barroso I, Luan J, Middelberg RP, Harding AH, Franks PW, Jakes RW, Clayton D, Schafer AJ, O’Rahilly S, Wareham NJ (2003) Candidate gene association study in type 2 diabetes indicates a role for genes involved in beta-cell function as well as insulin action. PLoS Biol 1(1):E20. doi:10.1371/journal.pbio.0000020
Jamshidi Y, Snieder H, Wang X, Pavitt MJ, Spector TD, Carter ND, O’Dell SD (2006) Phosphatidylinositol 3-kinase p85alpha regulatory subunit gene PIK3R1 haplotype is associated with body fat and serum leptin in a female twin population. Diabetologia 49(11):2659–2667. doi:10.1007/s00125-006-0388-z
Reiter V, Matschkal DM, Wagner M, Globisch D, Kneuttinger AC, Muller M, Carell T (2012) The CDK5 repressor CDK5RAP1 is a methylthiotransferase acting on nuclear and mitochondrial RNA. Nucleic Acids Res 40(13):6235–6240. doi:10.1093/nar/gks240
Liang Q, Li L, Zhang J, Lei Y, Wang L, Liu DX, Feng J, Hou P, Yao R, Zhang Y, Huang B, Lu J (2013) CDK5 is essential for TGF-beta1-induced epithelial-mesenchymal transition and breast cancer progression. Sci Rep 3:2932. doi:10.1038/srep02932
Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JR, Rayner NW, Freathy RM, Barrett JC, Shields B, Morris AP, Ellard S, Groves CJ, Harries LW, Marchini JL, Owen KR, Knight B, Cardon LR, Walker M, Hitman GA, Morris AD, Doney AS, Wellcome Trust Case Control C, McCarthy MI, Hattersley AT (2007) Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 316 (5829):1336–1341. doi:10.1126/science.1142364
Pascoe L, Tura A, Patel SK, Ibrahim IM, Ferrannini E, Zeggini E, Weedon MN, Mari A, Hattersley AT, McCarthy MI, Frayling TM, Walker M, Consortium R, Consortium UKTDG (2007) Common variants of the novel type 2 diabetes genes CDKAL1 and HHEX/IDE are associated with decreased pancreatic beta-cell function. Diabetes 56(12):3101–3104. doi:10.2337/db07-0634
Chang CL, Cai JJ, Huang SY, Cheng PJ, Chueh HY, Hsu SY (2014) Adaptive human CDKAL1 variants underlie hormonal response variations at the enteroinsular axis. PLoS ONE 9(9):e105410. doi:10.1371/journal.pone.0105410
Fuentes-Mattei E, Velazquez-Torres G, Phan L, Zhang F, Chou PC, Shin JH, Choi HH, Chen JS, Zhao R, Chen J, Gully C, Carlock C, Qi Y, Zhang Y, Wu Y, Esteva FJ, Luo Y, McKeehan WL, Ensor J, Hortobagyi GN, Pusztai L, Fraser Symmans W, Lee MH, Yeung SC (2014) Effects of obesity on transcriptomic changes and cancer hallmarks in estrogen receptor-positive breast cancer. J Natl Cancer Inst. doi:10.1093/jnci/dju158
Gong Z, Quan L, Yao S, Zirpoli G, Bandera EV, Roberts M, Coignet JG, Cabasag C, Sucheston L, Hwang H, Ciupak G, Davis W, Pawlish K, Jandorf L, Bovbjerg DH, Ambrosone CB, Hong CC (2013) Innate immunity pathways and breast cancer Risk in African American and European-American women in the Women’s Circle of Health Study (WCHS). PLoS ONE 8(8):e72619. doi:10.1371/journal.pone.0072619
Yao S, Graham K, Shen J, Campbell LE, Singh P, Zirpoli G, Roberts M, Ciupak G, Davis W, Hwang H, Khoury T, Bovbjerg DH, Jandorf L, Pawlish KS, Bandera EV, Liu S, Ambrosone CB, Zhao H (2013) Genetic variants in microRNAs and breast cancer risk in African American and European American women. Breast Cancer Res Treat 141(3):447–459. doi:10.1007/s10549-013-2698-4
Shifman S, Kuypers J, Kokoris M, Yakir B, Darvasi A (2003) Linkage disequilibrium patterns of the human genome across populations. Hum Mol Genet 12(7):771–776
Flegal KM, Shepherd JA, Looker AC, Graubard BI, Borrud LG, Ogden CL, Harris TB, Everhart JE, Schenker N (2009) Comparisons of percentage body fat, body mass index, waist circumference, and waist-stature ratio in adults. Am J Clin Nutr 89(2):500–508. doi:10.3945/ajcn.2008.26847
Katzmarzyk PT, Bray GA, Greenway FL, Johnson WD, Newton RL Jr, Ravussin E, Ryan DH, Smith SR, Bouchard C (2010) Racial differences in abdominal depot-specific adiposity in white and African American adults. Am J Clin Nutr 91(1):7–15. doi:10.3945/ajcn.2009.28136
Donohoe CL, Doyle SL, Reynolds JV (2011) Visceral adiposity, insulin resistance and cancer risk. Diabetol Metab Syndr 3:12. doi:10.1186/1758-5996-3-12
Catalan V, Gomez-Ambrosi J, Rodriguez A, Ramirez B, Andrada P, Rotellar F, Valenti V, Moncada R, Marti P, Silva C, Salvador J, Fruhbeck G (2015) Expression of S6K1 in human visceral adipose tissue is upregulated in obesity and related to insulin resistance and inflammation. Acta Diabetol 52(2):257–266. doi:10.1007/s00592-014-0632-9
Acknowledgments
This work was supported in part by grants from the US National Institutes of Health (P01 CA151135, R01 CA100598, K22 CA138563, and P30 CA072720), US Army Medical Research and Material Command (DAMD-17-01-1-0334), the Breast Cancer Research Foundation, and a gift from the Philip L. Hubbell family. The study used shared resources supported by Roswell Park Cancer Institute’s Cancer Center Support Grant from the National Cancer Institute (P30CA016056). The New Jersey State Cancer Registry is supported by the National Program of Cancer Registries of the Centers for Disease Control and Prevention under cooperative agreement 1US58DP003931-01 awarded to the New Jersey Department of Health. The collection of New Jersey cancer incidence data is also supported by the Surveillance, Epidemiology, and End Results program of the National Cancer Institute under contract N01-PC-2010-0027 and the State of New Jersey. The authors thank Dr. Dana Bovbjerg, Ms. Lina Jandorf, and Ms. Edie Prescod for their contribution to the Women’s Circle of Health Study. We also thank our research personnel at the Cancer Institute of New Jersey (now Rutgers Cancer Institute of New Jersey), Roswell Park Cancer Institute, Mount Sinai School of Medicine (now Icahn School of Medicine at Mount Sinai), UMDNJ School of Public Health (now Rutgers School of Public Health), and the New Jersey State Cancer Registry, as well as our African-American breast cancer advocates and community partners, and all the women who generously donated their time to participate in the study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cheng, TY.D., Shankar, J., Zirpoli, G. et al. Genetic variants in the mTOR pathway and interaction with body size and weight gain on breast cancer risk in African-American and European American women. Cancer Causes Control 27, 965–976 (2016). https://doi.org/10.1007/s10552-016-0774-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10552-016-0774-x