
Abstract
Purpose
HER2 gene is a member of the epidermal growth factor receptor (EGFR) family. Across different malignancies, aberrations of HER2 gene commonly correspond to gain-of-function alterations leading to increased receptor signaling.
Methods
We have reviewed the literature currently available on HER2 mutations in human breast cancer (BC) evaluating type and frequency of such mutations. The primary objective was to determine the frequency and the number of patients with HER2-mut in the series analyzed. The secondary objectives were to assess characteristics of mutated cases (ER and HER2 status and stage of disease, type of mutations, and finally the clinical outcome if reported).
Results
We retrieved 31 published papers, and the pooled rate of HER2 mutations across 12,905 BC patients was calculated. Overall, the frequency of HER2 mutations was 2.7% with most involving the intracellular domain. About 4% of patients were finally mutated. The predictive role was not described. Only 30% of these patients were simultaneously HER2 positive and 63% were ER positive.
Conclusion
We have found that the prevalence of HER2 mutations is about 3%. These genic alterations are independently associated with HER2 amplification status, occurring in both ER-positive/HER2-negative diseases or HER2-enriched cancers. Ongoing trials are investigating small molecules tyrosine kinase inhibitors in patients harboring these mutations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The human epidermal growth factor receptor 2 (HER2) gene encodes for a 185-KD transmembrane glycoprotein receptor with an intracellular domain with tyrosine kinase activity. The HER2 receptor belongs to the epidermal growth factor receptors’ family, who are responsible for the activation of intracellular signal transduction pathways controlling epithelial cell growth, differentiation, and angiogenesis [1,2,3,4,5]. Amplification of HER2 or overexpression of its protein product is observed in 18–20% of human breast cancers (BCs) and identify a subgroup of women who can benefit from treatment with agents targeting HER2, such as trastuzumab, both in adjuvant and in metastatic settings [6,7,8]. Unfortunately, a proportion of HER2-positive BC patients either recur after adjuvant trastuzumab or progress during systemic therapy for advanced disease showing resistance (primary or acquired) to anti-HER2 treatment. Different mechanisms have been associated with reduced efficacy of trastuzumab in vitro. These include aberrations in HER2 signaling as expression of the truncated HER2 receptor fragment p95; activating mutations of phosphatidylinositol 3-kinase (PIK3CA) gene; loss of phosphatase and tensin homolog (PTEN) gene; activation of other downstream signal transducers; prevention of cell cycle arrest; increased signaling through alternative tyrosine kinase receptors; and resistance to antibody-dependent cellular cytotoxicity (ADCC) [9]. In clinical practice, data of drug resistance derive from small case series or retrospective analysis of randomized studies, and this represents insufficient evidence to inform for patients’ selection.
Some rare mutations in tyrosine kinase domain of HER2 protein (e.g., exon 19 and 20) resulted in a more potent receptor form than wild-type HER2 (HER2-wt) in activating signal transducers, phosphorylating EGFR, and inducing survival, invasiveness, and tumorigenicity, with reduced sensitivity to anti-EGFR agents but retained sensitivity to anti HER2 agents as lapatinib [10]. These mutated variants showed an increased tyrosine kinase activity compared with wild-type counterpart. Furthermore, secondary lapatinib resistance may develop due to kinase domain mutations in preclinical models. In these cases, irreversible HER2 inhibitors may offer alternative options to breast cancer and other solid tumor patients harboring lapatinib resistance mutations [11]. In addition, most of these mutations rise in HER2-negative (nonoverexpressed or nonamplified) samples [12]. However, few data are currently available from prospective trials in human BCs.
In order to evaluate the frequency, the type, and the significance of HER2 mutations (HER2-mut) in human BC, we have performed a systematic review of the literature on this topic.
Materials and methods
Electronic search and study selection
An electronic search in Pubmed (“receptor, erbb-2”[MeSH Terms] OR (“receptor”[All Fields] AND “erbb-2”[All Fields]) OR “erbb-2 receptor”[All Fields] OR “her 2”[All Fields] OR “genes, erbb-2”[MeSH Terms] OR (“genes”[All Fields] AND “erbb-2”[All Fields]) OR “erbb-2 genes”[All Fields]) OR (““receptor, erbb-2”[MeSH Terms] OR (“receptor”[All Fields] AND “erbb-2”[All Fields]) OR “erbb-2 receptor”[All Fields] OR “erbb2”[All Fields] OR “genes, erbb-2”[MeSH Terms] OR (“genes”[All Fields] AND “erbb-2”[All Fields]) OR “erbb-2 genes”[All Fields])) AND (“:breast neoplasms”[MeSH Terms] OR (“breast”[All Fields] AND “neoplasms”[All Fields]) OR “breast neoplasms”[All Fields] OR (“breast”[All Fields] AND “cancer”[All Fields]) OR “breast cancer”[All Fields]) AND (“mutation”[MeSH Terms] OR “mutation”[All Fields]), EMBASE. and The Cochrane Library was performed from inception up to February 18, 2017. All the resulting studies were retrieved, and their cited references simultaneously checked for other potentially relevant publications. Review articles and related articles found on Pubmed were also scanned to find additional eligible studies. For studies on the same population of patients, only the more updated published one was selected. The language of publication was restricted to English.
Data extraction and statistical analysis
The primary objective was to determine the frequency and number of patients with HER2-mut in the series analyzed. The secondary objectives were to assess characteristics of mutated cases (ER and HER2 status and stage of disease, type of mutations, and finally the clinical outcome if reported). Author, year, type of study, stage, and the number of screened patients were primarily extracted. Also, data on the number (rate) of mutated BCs, site of mutations, and pathological characteristics of mutated BCs were retrieved. Data were pooled and presented with descriptive statistics using Medcalc statistical software version 17.2 (MedCalc Software bvba, Ostend, Belgium; http://www.medcalc.org; 2017).
Results
Based on initial search strategy, 2901 studies were screened. Among them, 31 were deemed eligible for inclusion in the final analysis. See Table 1 for characteristics of included studies. Most studies were retrospective series (n = 26; range 22–5605) o single case reports (n = 5). Median number of patients analyzed was n = 82.
Description of studies and of mutated patients
The included studies evaluated a total of 12,905 BC patients. Subjects included were mainly early stage BCs, but metastatic patients were also included. Median-pooled ER-positive and HER2-positive in HER2 mutated BCs were 78.6 (pooled mean, 63.6%) and 0% (pooled mean, 30%), respectively.
Pooled rate of mutated breast cancer and rate of mutations
HER2 mutations were detected in 338 patients of the total population (pooled proportion 3.9%; 95% CI 2.9–5%; I 2 = 82.36%, random-effects model) with different stages of disease (Fig. 1). The number of mutations was 356 (prevalence 2.7%). Mutation/patient rate was 1.05.

Pooled proportion of HER2-mutated breast cancers in all included patients
Type and site of mutations
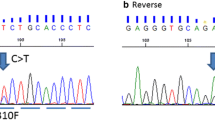
Mutations were all characterized and described by authors in either full text or supplemental files except for n = 2. Type and frequency of mutations were included in Table 2 and Fig. 2. More than 50% (n = 189) concern two main domains: the TKI domain (n = 163 mutations; L755S, V777L, and D769H or D769Y), and the Furin like domain (n = 26; S310F and S310Y). The most frequent was the missense L755S (Mutation Id: Cosm14060) mutation (n = 86; 24%) that is associated to resistance to lapatinib and response with neratinib in preclinical studies, but did not produce oncogenic transformation (Bose et al.). It is located in the exon 19 and results in an amino acid substitution at position 755 in HER2, from a leucine (L) to a serine (S). The second-most frequent activating mutation was the missense V777L (n = 49; 13.7%) that is associated to response to trastuzumab, lapatinib, and neratinib in preclinical studies (Mutation Id: Cosm14062). It is located in the exon 20 and results in an amino acid substitution at position 777 in HER2, from a valine (V) to a leucine (L). Finally, the third more frequent somatic mutations are the D769H and D769Y missense-activating mutations (n = 28; 7.8%) that are both associated to response to trastuzumab, lapatinib, and neratinib in preclinical studies. The D769H (Mutation Id: 13170) is located in the exon 19 and results in an amino acid substitution at position 769 in HER2, from an aspartic acid (D) to a histidine (H). The D769Y mutation (Mutation Id: 1251412) results in an amino acid substitution at position 769 in HER2, from an aspartic acid (D) to a tyrosine (Y), and is located in exon 19. Finally the L755_T759del in-frame deletion mutation (Mutation Id: 029269) occurs in HER2 in the kinase domain of exon 19 and is associated to lapatinib resistance in preclinical models. It represents the 1.6% of our series. It is known for increasing phosphorylation of HER2’s heterodimerization partners: EGFR and HER3 (Bose et al.).

Rates of mutations recorded with >1% of frequence
Correlation with outcome o response
Correlation of mutations with response to anti-HER2 agents were seldom reported in single case reports. In two large series (Zuo et al. and Wang et al.), correlation with outcome was presented. In Zuo et al., BC with HER2 mutations exhibited decreased relapse-free survival (P = 0.02) compared with breast cancer bearing HER2 wild type. In Wang et al., all subjects with a HER2 somatic mutation had a significantly worse outcome and, in particular, those with HER2-negative BC: reduced recurrence-free survival (unadjusted hazard ratio [HR] 2.67; 95% confidence interval [CI] 1.25–5.72, P = 0.002), and distant recurrence-free survival (unadjusted HR 2.50; 95% CI 1.10–5.68, P = 0.004) than those with wild-type HER2 status.
Discussion
In some tumor types (e.g., breast and gastroesophageal), aberrant proliferation signaling is caused by HER2 receptor overexpression and/or gene amplification, or by elevated levels of the ligands. More recently, the activating mutations resulting in constitutive functioning of the tyrosine kinase activity domain have been described. The HER2 receptor is a potential site of mutations that are clustered mainly in two different spots: the extracellular domain at exon 8 and the kinase domain at exons 19 and 20. However, mutations can also occur in other regions of the receptor. At present, the real frequency in clinical practice, the therapeutic options and the clinical meaning of these rare mutations are unknown. Hence, we have performed a systematic search of the literature to address this issue focusing on human BC.
Across 31 retrospective series or case reports, we evaluated the prevalence of HER2 mutations among 12,905 BC patients. We identified 356 mutations in 338 patients (prevalence of patients with HER2 mutations 2.7%). The most represented mutations were L755S (24%), V777L (13.7%), and DF769H or Y (7.8%) that collectively comprised 151 mutations (53% of the total amount). All these three types of mutations are activating mutations, and, except the first, the other two are associated with sensitivity to trastuzumab, lapatinib, and neratinib. Expressions of estrogen receptor and HER2 status were variable (most were ER positive, only 30% were HER2 positive). Data on the correlation of HER2 mutations with outcome were scarce, with some reports showing an association with other mutations (e.g., p 53) and others reporting associations with lobular invasive carcinoma. In general, the prognosis was poor compared with HER2-w.t counterpart.
Preclinical evidence demonstrated that a subset of somatic HER2 mutations are activating (gain-of-function) mutations that retain sensitivity or resistance to selective HER2 tyrosine kinase inhibitors, but data in clinical practice are formally lacking. In particular, the activity of the available agents used upfront (e.g., trastuzumab or pertuzumab) in the presence of these mutations is not well established. In a next-generation sequencing (NGS) analysis of 7300 tissue samples from 403 different tumors, Chmielecki et al. found oncogenic HER2 mutations in about 32% of these tumors [12]. Some mutations, such as V777L, are clearly activating mutations because they are strongly associated with the increased phosphorylation of signaling proteins. The HER2 mutation L755S has been associated with resistance to lapatinib, but also sensitivity to neratinib as demonstrated by Bose et al. and Kancha et al. [11, 13]. Specifically, leucines at L755 participate in hydrophobic interactions of the activation loop, and the substitution of leucine with serine at this point is expected to destabilize to an inactive form, being the conformation required for lapatinib binding. Xu and colleagues demonstrated also that HER2 reactivation through acquisition of the L755S mutation was a mechanism of acquired resistance to lapatinib-based therapy in preclinical HER2-amplified BC cell lines, which can be overcome by irreversible HER1/2 inhibitors [14].
Some mutations in individual domains that may have direct effects on downstream signaling may also have therapeutic implications, as anticipated above. In some cases, in fact, actionable alterations on drug targets can affect drug efficacy. For example, S310F/Y mutations sensitize cancer cell lines to the tyrosine kinase inhibitors neratinib, afatinib, and lapatinib [15]. A mutation in tyrosine kinase domain named T798I, is functionally similar to T790M mutation of EGFR, which is known to be associated with resistance to erlotinib and gefitinib in non–small cell lung cancers (NSCLCs). In fact, ErbB2 T798I-expressing cells survive and grow in the presence of high lapatinib concentrations [16]. Among mutations associated with resistance to lapatinib, the only other mutation found (n = 1; 0.3% of all mutations analyzed) was the L726F. In such a mutation, the aliphatic side chain of leucine is replaced with the rigid, bulkier phenylalanine side chain.
Other authors have found that HER2 missense mutations are functionally distinct and require additional genetic alterations like PIK3CA mutation in V777L, to promote features of malignant transformation [17]. Therefore, missense mutations by themselves may not be reliable negative predictors of response to HER2-targeted therapies. A recent meta-analysis showed that PIK3CA mutations are not associated with inferior benefit in early and metastatic BC patients treated with trastuzumab [18]. Only in the neoadjuvant setting, patients harboring wild-type PIK3CA tumors attained a statistically significant higher pathologic complete response (pCR) rate.
HER2 mutations have been reported in lung, gastrointestinal, gynecological, and genitourinary cancers [12]. In HER2-mutated lung adenocarcinoma, a similar frequency of HER2 amplification and mutations (3%) was found, but none of the HER2-mutant cases was amplified, thus suggesting diseases with a distinct molecular profile and possibly therapeutic target [19]. Wen et al. analyzed 7497 specimens from a variety of solid tumors and found HER2 kinase domain mutations in approximately 1% of all cases, ranging from absent in sarcomas to 4% in urothelial cancers [20]. Most of these were activating mutations, and about 20% of cases had coexisting HER2 amplification and/or overexpression. Interestingly, the clinical characteristics of 65 out of 3800 patients (1.7%) discovered as carriers of HER2 in-frame insertion in exon 20 were recently described [21]. All tumors were adenocarcinomas, and most of them affected women who did not have a smoking history. Half of the patients were metastatic. Among these stage IV cases, 16 were treated with anti-HER2 therapies, and reported a disease control rate ranging from 93% with trastuzumab to 100% with afatinib. Inducible tissue-specific overexpression of the most common mutant HER2 (HER2(YVMA)) in mouse models was found to result in rapid development of NSCLC, confirming the oncogenicity of this driver mutation [22]. A phase I study evaluated the combination of neratinib and temsirolimus in patients with advanced solid tumors, and some level of clinical activity was reported in HER2-mutant NSCLC [23]. Furthermore, in bladder cancer, HER2-activating mutations were recently found to be associated with an excellent rate of pCR (approximately 24%) after neoadjuvant chemotherapy [24].
Some phase II studies are currently ongoing, recruiting patients with HER2-mutated BC. One study is evaluating neratinib alone or in combination with fulvestrant in ER-positive BC with overall response rate as primary endpoint (NCT01670877). Another study is investigating poziotinib (a panHER tyrosine kinase inhibitor) in patients with HER2- or EGFR-mutated BC (NCT02544997). Finally, the PUMA-NER study (NCT01953926) is evaluating response rates at week 8 in patients with EGFR/HER2- or HER3-mutated solid tumors. Notably, in a preliminary analysis of the basket SUMMIT phase II trial testing neratinib in nonamplified HER2 tumors harboring activating HER2 mutations, an interesting 32% objective response rate with a 3.5 months median PFS was reported in 25 patients with HER2-mutated BC [25]. The interim safety results of the study showed that the most frequently observed adverse event was diarrhea (22% grade 3 in the overall population), and thanks to the prophylactic use of loperamide, its duration was short (median 2 days) and did not represent a treatment-limiting side effect.
This work portends some intrinsic limitations. First, this is a retrospective analysis of HER2 mutations in a series of different patient populations (initial stages and advanced disease, HER2-negative and -positive BCs, different methods for searching mutations); hence, results cannot be generalized to current clinical practice. In particular, the clinical characteristics of HER2 mutation carriers cannot be defined. Second, details on the treatment received by these subjects are lacking; therefore, eventual benefit with specific therapies is unknown. Finally, the outcome was generally not reported, so whether HER2 mutations were associated with poorer prognosis compared with the wild-type counterpart is presently uncertain. To the best of our knowledge, these data represent, however, the first systematic analysis on frequency and type of HER2 mutations in about 13,000 BCs across diseases in different stages. Results of our research reveal that HER2 mutations are relatively rare in human BCs (2.7%) and in large part not related to HER2-neu gene amplification. Among 356 mutations recorded from 231 publications or case reports, we found that the most frequent mutations occur in the tyrosine kinase intracellular domain and are either associated with constitutive kinase activity with consequent oncogenic transformation of normal cells (i.e., V777L) but still sensitivity to anti-HER2 agents, or not associated with transforming potential (i.e., L755S), but showing resistance to lapatinib. Although rare, we believe that the eventual presence of these mutations should always be considered when treating advanced refractory BCs, and a specific search for such mutations should be offered to patients for possible treatment with available targeted drugs. Screening for HER2 mutations could be hypothesized in HER2-positive BC patients progressing during lapatinib treatment, but could be searched even in luminal BC, after exposure to all labeled agents and when no more active options are available at best. Otherwise, two-thirds of patients carrying HER2 mutations are HER2 negative (70%) or ER+ (80%); therefore, given the worst outcome of HER2-negative BC patients who carry a HER2 somatic mutation, they are potential candidates for receiving HER2-targeted therapy plus chemo- or endocrine therapy, or for recruitment into ongoing clinical trials. It is well known that metastatic BC if re-biopsed, could show a shift of HER2 expression from initially positive into negative, and therefore it is reasonable to perform HER2 testing in those metastatic sites accessible for biopsy. Bearing in mind the HER2 mutations data here presented, it could be also advisable to verify if any HER2-mutated variants, in distant metastases, occurred. In addition, a crosstalk between ER and HER2 has been described as a mechanism of resistance to endocrine therapy. The presence of hyperactive growth factor receptor signaling, as occurs in HER2 overexpressed (or amplified) BC, possibly leads to an excessive phosphorylation of ER and its coregulators, reducing the inhibitory effects of hormonal therapies. Similarly, HER2 mutations are oncogenic drivers in HER2-negative BC and represent the rationale for the dual blockade of ER and HER2 signaling to enhance their antitumor activity in HER2-negative but mutated metastatic BC.
In conclusion, our data indicate that in BCs, the prevalence of HER2 mutations is about 3%. These genic alterations are independently associated with HER2-amplification status, occurring in both ER-positive/HER2-negative diseases or HER2-enriched cancers. These findings may have important clinical implications thanks to the possibility to avoid potential ineffective therapies in salvage setting and to screen for potential new therapeutic targets and alternative treatment options.
References
King CR, Kraus MH, Aaronson SA (1985) Amplification of a novel v-erbB-related gene in a human mammary carcinoma. Science 229:974–976
Karunagaran D, Tzahar E, Beerli RR, Chen X, Graus-Porta D, Ratzkin BJ et al (1996) ErbB-2 is a common auxiliary subunit of NDF and EGF receptors: implications for breast cancer. EMBO J 15:254–264
Petit AM, Rak J, Hung MC, Rockwell P, Goldstein N, Fendly B et al (1997) Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases down-regulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: angiogenic implications for signal transduction therapy of so. Am J Pathol 151:1523–1530
Klapper LN, Glathe S, Vaisman N, Hynes NE, Andrews GC, Sela M et al (1999) The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc Natl Acad Sci USA 96:4995–5000
Giatromanolaki A (2004) c-erbB-2 related aggressiveness in breast cancer is hypoxia inducible factor-1 dependent. Clin Cancer Res 10:7972–7977
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235:177–182
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A et al (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344:783–792
Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, Smith I et al (2005) Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med 353:1659–1672
Luque-Cabal M, García-Teijido P, Fernández-Pérez Y, Sánchez-Lorenzo L, Palacio-Vázquez I (2016) Mechanisms behind the resistance to trastuzumab in HER2-amplified breast cancer and strategies to overcome it. Clin Med Insights Oncol 10:21–30
Wang SE, Narasanna A, Perez-Torres M, Xiang B, Wu FY, Yang S et al (2006) HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 10:25–38
Kancha RK, von Bubnoff N, Bartosch N, Peschel C, Engh RA, Duyster J (2011) Differential sensitivity of ERBB2 kinase domain mutations towards lapatinib. PLoS ONE 6:e26760
Chmielecki J, Ross JS, Wang K, Frampton GM, Palmer GA, Ali SM et al (2015) Oncogenic alterations in ERBB2/HER2 represent potential therapeutic targets across tumors from diverse anatomic sites of origin. Oncologist 20:7–12
Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC et al (2013) Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov 3:224–237
Xu X, De Angelis C, Burke KA et al (2017) HER2 Reactivation through acquisition of the HER2 L755S mutation as a mechanism of acquired resistance to HER2-targeted therapy in HER2(+) breast cancer. Clin Cancer Res
Greulich H, Kaplan B, Mertins P, Chen T-H, Tanaka KE, Yun C-H et al (2012) Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc Natl Acad Sci USA 109:14476–14481
Trowe T, Boukouvala S, Calkins K, Cutler RE, Fong R, Funke R et al (2008) EXEL-7647 inhibits mutant forms of ErbB2 associated with lapatinib resistance and neoplastic transformation. Clin Cancer Res 14:2465–2475
Zabransky DJ, Yankaskas CL, Cochran RL, Wong HY, Croessmann S, Chu D et al (2015) HER2 missense mutations have distinct effects on oncogenic signaling and migration. Proc Natl Acad Sci USA 112:E6205–E6214
Ibrahim EM, Kazkaz GA, Al-Mansour MM, Al-Foheidi ME (2015) The predictive and prognostic role of phosphatase phosphoinositol-3 (PI3) kinase (PIK3CA) mutation in HER2-positive breast cancer receiving HER2-targeted therapy: a meta-analysis. Breast Cancer Res Treat 152:463–476
Li BT, Ross DS, Aisner DL, Chaft JE, Hsu M, Kako SL et al (2016) HER2 amplification and HER2 mutation are distinct molecular targets in lung cancers. J Thorac Oncol 11:414–419
Wen W, Chen W, Xiao N, Bender R, Ghazalpour A, Tan Z et al (2017) Mutations in the kinase domain of the HER2/ERBB2 gene identified in a wide variety of human cancers. J Mol Diagnostics 17:487–495
Mazières J, Peters S, Lepage B, Cortot AB, Barlesi F, Beau-Faller M et al (2013) Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol 31:1997–2003
Perera SA, Li D, Shimamura T, Raso MG, Ji H, Chen L et al (2009) HER2YVMA drives rapid development of adenosquamous lung tumors in mice that are sensitive to BIBW2992 and rapamycin combination therapy. Proc Natl Acad Sci USA 106:474–479
Gandhi L, Bahleda R, Tolaney SM, Kwak EL, Cleary JM, Pandya SS et al (2017) Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2-dependent and other solid tumors. J Clin Oncol 32:68–75. doi:10.1200/JCO.2012.47.2787
Groenendijk FH, de Jong J, Fransen van de Putte EE, Michaut M, Schlicker A, Peters D et al (2016) ERBB2 mutations characterize a subgroup of muscle-invasive bladder cancers with excellent response to neoadjuvant chemotherapy. Eur Urol 69:384–388
Hyman DM, Piha-Paul SA, Rodon J et al. Abstract CT001: Neratinib in HER2 or HER3 mutant solid tumors: SUMMIT, a global, multi-histology, open-label, phase 2” basket” study
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Petrelli, F., Tomasello, G., Barni, S. et al. Clinical and pathological characterization of HER2 mutations in human breast cancer: a systematic review of the literature. Breast Cancer Res Treat 166, 339–349 (2017). https://doi.org/10.1007/s10549-017-4419-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-017-4419-x