
Abstract
Biomarkers of bone turnover, including urine N-telopeptide (uNTx), have been used as surrogate measures of response to bone-targeted therapies. Vascular endothelial growth factor (VEGF) levels correlate with extent of bone metastases. We assessed whether vandetanib, an inhibitor of VEGF, epidermal growth factor receptor and RET signalling, improved uNTx response when added to fulvestrant (F) in breast cancer patients with bone metastases. Postmenopausal patients with bone predominant, hormone-receptor-positive metastatic breast cancer were randomised to F (500 mg IM days 1, 15, 29, then monthly) with either vandetanib (100 mg PO OD) (FV) or placebo (FP). The primary objective was uNTx response. Secondary objectives included PFS, OS, RECIST response, pain scores and toxicity. Sixty-one patients were allocated to FV and 68 to FP. Out of 127 analyzable patients, an uNTx response occurred in 66 % for FV and 54 % for FP (p = 0.21). No difference was detected between groups for PFS; HR = 0.95 (95 % CI 0.65–1.38) or OS HR = 0.69 (95 % CI 0.37–1.31). For the 62 patients with measurable disease, clinical benefit rates were 41 and 43 %, respectively (p = 0.47). Serious adverse events were similar, 3.3 % for FV versus 5.9 % for FP. Elevated baseline uNTx (>65 nM BCE/mmol Cr) was prognostic for PFS, HR = 1.55 (95 % CI 1.04–2.30) and for OS, HR = 2.32 (95 % CI 1.25–4.33). The addition of vandetanib to fulvestrant did not improve biomarker response, PFS or OS in patients with bone metastases. Baseline bone turnover was prognostic for PFS and OS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer patients with bone metastases limited to the skeleton frequently have indolent disease with a prolonged disease course. Our increased understanding of the interaction between cancer cells in bone and stromal elements such as osteoclasts has led to the extensive use of bone-targeted agents such as bisphosphonates and RANKL antibodies for patients with bone metastases. To date, these therapies have had no impact on either progression free or overall survival (OS) [1]. In patients with bone metastasis, vascular endothelial growth factor (VEGF) levels correlate with extent of bone disease [2] and suppression of VEGF concentrations are associated with improved outcomes [3]. In addition, members of the VEGFR receptor family (VEGFR-1, VEGFR-2 and VEGFR-3) [4] and epidermal growth factor receptor (EGFR) signalling pathways are recognised targets for breast cancer therapy.
Vandetanib (AstraZeneca, Macclesfield, UK) is a once-daily oral agent that targets VEGFR and EGFR signalling, [5] as well as RET (rearranged during transfection) tyrosine kinase [6]. RET tyrosine kinase is involved in breast tumour growth and endocrine resistance [7, 8]. Vandetanib has shown anti-tumour activity both as a single agent or in combination therapy in a number of malignancies including non-small-cell lung cancer [9] and medullary thyroid cancer [10]. Vandenib has been evaluated both as a single agent [11, 12] and in combination with chemotherapy [13] in patients with refractory metastatic breast cancer (MBC).
Breast cancers that are metastatic to bone are often hormone-receptor-positive and, in postmenopausal women with metastatic disease, first and second line endocrine therapies frequently include aromatase inhibitors (AI) and tamoxifen. The oestrogen receptor down regulator, fulvestrant, has demonstrated activity after both tamoxifen and AI [14].
We hypothesised that the use of agents which target VEGF and EGFR signalling could potentially enhance the effects of fulvestrant in inhibiting the growth of bone metastases. We, therefore, conducted a randomised, double-blind, multicentre, phase II study (ZAMBONEY; NCT00811369) to investigate whether the addition of once-daily oral vandetanib to fulvestrant could reduce bone resorption in postmenopausal patients with bone only or bone predominant metastatic, hormone-receptor-positive breast cancer.
Patients and methods
Patients
The main inclusion criteria were postmenopausal woman with either radiologically confirmed bone only or predominant metastases to bone and an oestrogen -receptor-positive (ER+) and/or progesterone-receptor-positive (PgR+) primary tumour. Patients’ disease had to fulfil one of the following criteria for endocrine resistance: (i) disease progression on tamoxifen or on an AI as first or second line therapy for metastatic disease or (ii) development of metastatic disease while on treatment with tamoxifen or an AI in the adjuvant setting or (iii) disease progression after discontinuation of prior adjuvant endocrine therapy. Patients additionally had to have bone lesions which were lytic, sclerotic or mixed, either in the absence or the presence of measurable disease as defined by response evaluation criteria in solid tumours (RECIST) criteria. Determination of 'bone predominant' disease was left up to the treating investigator. This was not centrally determined or adjudicated.
Exclusion criteria included: anticipated life expectancy less than 6 months, previous treatment with fulvestrant or vandetanib, having received greater than one line of palliative chemotherapy, significant cardiovascular event within 3 months of study entry or a history of symptomatic arrhythmias. In addition, patients with a history of congenital long QT syndrome, QT prolongation or taking medications with a risk of QTc prolongation were excluded. While prior treatment with VEGF TKIs was a study exclusion, prior use of bevacizumab was permitted. Prior and continuing bisphosphonate use was permitted as long as this was commenced at least a month prior to study entry.
Study design and treatments
This trial was conducted in 13 Canadian cancer centres. Patients provided written informed consent and the trial was approved by all local research ethics committees. Patients were randomly assigned (1:1) to receive vandetanib (100 mg/day) plus fulvestrant (500 mg IM days 1, 15, 29 and every 28 days afterwards) or to placebo plus fulvestrant using a web-based subject registration and randomization system located at the Ontario Clinical Oncology Group (OCOG) Coordinating and Methods Centre. A computer-generated randomization schedule was used to allocate patients using variable block sizes, within strata defined by baseline fasting serum C-telopeptide (CTx) level (<400 ng/L, ≥400 ng/L) and clinical center. Active and placebo tablets were identical. Study treatment was to continue until disease progression, unacceptable toxicity or withdrawal of consent. Toxicity-related dose adjustments were performed in accordance with protocol-specified guidelines. Data and study management were coordinated through OCOG.
Radiological assessments
Radiological tumour assessments were performed at baseline and repeated every 12 ± 2 weeks until either disease progression or treatment intolerance. For patients with measurable disease, radiological response was defined as per RECIST Version 1.0 [15].
Laboratory studies
For patient stratification, Serum CTx (β-CrossLaps) for study stratification was drawn in the morning following an overnight fast. The sample was allowed to clot, and centrifuged at 4 °C for 10 min at 3,400 revolutions per minute. Aliquots of serum were shipped immediately on dry ice to the central lab (Mount Sinai Hospital, Toronto) for analysis. Serum CTx was measured by chemiluminescence immunoassay using CrossLaps®. Intra-individual variance is less than 18 %, and the coefficient of variation is <5 %. Specimens for urinary NTx were collected as second pass specimens after an overnight fast and stored in a −30 to −70°C freezer until batch shipped to the central lab. Urine NTx was measured by competitive-inhibition enzyme-linked immunosorbent assay (ELISA). Assay range is from 20–3,000 nmol/L BCE, with intra-assay variability of 7.6 % and inter-assay variability of 4 %.
Outcomes
The primary outcome of a significant change in the bone turnover marker urinary N-telopeptide (uNTx) level was defined as a ≥30 % reduction in uNTx level from baseline at any time. This definition was used by Vinholes et al. [16] and was the approximate magnitude observed in recent salvage bisphosphonate trials [17]. No minimum baseline level of uNTx was mandated for study inclusion.
Secondary outcomes included progression-free survival (PFS), defined as the time from randomization until disease progression or death. Progression was defined according to RECIST criteria [18] (where applicable), or for patients with lytic or mixed bone lesions and no measureable disease, as the appearance of one or more new lytic lesions in bone, the appearance of one or more new lesions outside of bone or unequivocal progression of existing bone lesions. PFS was measured locally, and no central adjudication of disease progression was performed. Additional secondary outcomes included response to therapy according to RECIST criteria, OS (calculated from the date of randomization to the date of death by any cause), pain response as measured by the Brief Pain Inventory (BPI) [19] and FACT-BP [20] questionnaires, and number of study on skeletal-related events (SREs). Safety was assessed by recording adverse events using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE), Version 3.0. The study was closed on 31 July 2013 on the basis of the Data Safety Monitoring Committee recommendation. All patients still receiving treatment at this time were censored for PFS and OS.
Statistical considerations
Based on our previous phase II study which evaluated urinary markers in a similar group of patients, we postulated that 15 % of patients on fulvestrant alone would have a major (>30 %) drop in uNTx and with the addition of vandetanib as many as 40 % of patients receiving combination therapy would have a drop in their urinary NTx relative to baseline [9]. With alpha = 5 % (two sided), power of 80 %, 57 patients per group were needed to detect a difference of this magnitude using a Fisher’s exact test. Assuming a conservative dropout rate of 10 %, total accrual was set to 126 patients.
Although the protocol-defined primary analysis was based on Fisher’s exact test, the Mantel–Haenszel (M–H) test was also performed accounting for the CTx classification at baseline. For the secondary outcomes, treatment arms were compared using Fisher’s exact tests, M-H tests, Wilcoxon rank sum tests and using regression (logistic, Cox, linear) models to adjust for strata. PFS and OS were estimated using the Kaplan–Meier method. Baseline serum CTx and uNTx were evaluated as potential prognostic and predictive (testing for an interaction) factors for PFS and OS using Cox regression analyses. Whether a change in urine NTx by week 4 was prognostic for PFS and OS was also tested using a landmark analysis. Baseline urine NTx was defined using a continuous variable with a logarithmic transformation (for statistical normalisation purposes), and using a definition of normal versus abnormal (>65 nM BCE/mmol Cr). Serum CTx was defined using the baseline stratification of <400 versus ≥400. All tests were two-sided and a p value of 0.05 or less was considered statistically significant. Analyses were performed in SAS v9.2 (Cary, NC) and R (www.r-project.org).
Results
Patients
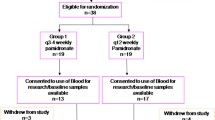
Between October 2009 and October 2011, 133 patients were registered. Three of these patients were withdrawn prior to randomization due to physician discretion, and one patient was randomised in error (to FV) and subsequently withdrawn prior to receiving any treatment. These patients are excluded from all remaining analyses (Fig. 1). Of the remaining 129 patients, 61 were assigned to receive FV and 68 to receive FP. All but 3 patients received fulvestrant and vandetanib/placebo within one day of randomization; one patient started treatment 29 days after randomization, a second patient 4 days after randomization, and one patient (FV arm) withdrew and transferred their care to another institution prior to receiving any treatment. One patient did not have baseline uNTx measured (FV arm) but did have all post-baseline uNTx measurements.

Study flow diagram
Baseline characteristics
Table 1 shows the baseline patient characteristics. Median age was 59, 18 % had received 1 prior chemotherapy regimen and 73 % prior endocrine therapy for MBC. The women who received FV were less likely to have had measurable disease by RECIST criteria (34.4 vs 58.8 %) and less likely to have had prior chemotherapy (9.9 vs 25.0 %) for metastatic disease.
Primary outcome, urinary NTx response
Of the 59 women treated with FV and having baseline uNTx, 39 (66.1 %) had a decline of 30 % or more in uNTx, compared with 37 of 68 (54.4 %) of women in the FP arm (difference = 11.7 %; 95 % confidence interval (CI) −5.2 to 28.6 %, Fisher’s exact p = 0.21, M–H p = 0.10). The maximum decrease in uNTx at any time while on-treatment for all patients is shown in a waterfall plot (Fig. 2).

Waterfall plot of percentage change in urine NTx from baseline
No statistically significant difference between treatment arms was observed for any supportive analysis using different definitions for the change in uNTx. This includes analyses where only second pass urine samples were included, using a drop of 50 % or more as the definition of response, whether a drop occurred within the first 4 weeks of treatment, and when examining the change in uNTx from baseline as a continuous measure at selected time points.
Secondary outcomes
Median PFS was 5.8 months (95 % CI 2.7–8.1) and 4.8 months (95 % CI 2.7–5.4), respectively, for FV and FP (p value = 0.73, HR = 0.94 (95 % CI 0.64–1.36, Fig. 3a). For those patients with measurable disease, clinical benefit rates were 41 and 43 %, respectively. At the time of study closure, 41 women had died, 18 in the FV treatment arm and 23 on FP. Two-year OS was 64.6 % (95 % CI 43.7–79.4 %) and 60.6 % (45.5–72.7 %) for FV and FP patients (p = 0.30, Fig. 3b). No statistically significant difference in any outcome including on-study SREs (Table 2) or FACT-BP pain scores (Table 3) was observed. Statistically significant differences in BPI average pain and severity scores were observed.

a Progression-free survival and b overall survival by treatment and baseline urinary NTx
Safety
Fourteen (23.0 %) patients on FV and 9 (13.2 %) on FP experienced a grade 3 or greater serious or clinically significant AE (Fisher’s exact test p value = 0.17). Six patients experienced a serious adverse event, 2 who received FV (diarrhoea and mucositis/stomatitis) and 4 who received FP (dehydration, pericarditis and 2 cases of pulmonary infection).
Exploratory analyses
Cox proportional hazards models, adjusted for treatment and baseline CTx, were used to assess differences in pre-defined subgroups of hormone sensitive disease (prior adjuvant endocrine therapy for over 24 months or a prior line of palliative endocrine therapy for over 24 weeks) and site of disease (bone only vs other). No significant effect was observed for hormone sensitivity (PFS p value = 0.25 and OS p value = 0.88), however, patients with bone only disease had significantly improved median PFS (median = 7.9 vs 2.8 months, p value = 0.006) and OS (2-year OS = 80.6 vs 44.2 %, p value = 0.003) compared to patients with additional metastatic disease outside of the skeleton.
Baseline uNTx and serum CTx were statistically significantly prognostic for PFS and OS. Across treatments, patients with abnormal baseline uNTx (>65 nM BCE/mmol Cr) had a 1.51 (95 % CI 1.02–2.25, p = 0.041) increased hazard of PFS and a 2.42 (95 % CI 1.31–4.48, p = 0.005) increased hazard of dying. An interaction between treatment arm and baseline uNTx was also observed (p = 0.028) with PFS (see Fig. 3a). Women with normal baseline uNTx had similar median PFS regardless of treatment (5.4 months, 95 % CI 2.7–8.0 for FV vs 5.4 months, 95 % CI 3.8–11.3 for FP). However, amongst women with abnormal baseline uNTx, those women who received FV had improved median PFS (7.4 months, 95 % CI 2.4–10.9 for FV vs 2.5 months, 95 % CI 2.4–2.8 for FP). A similar trend was observed (see Fig. 3b) for OS; however, the interaction term was not statistically significant (p = 0.25).
Interestingly, the percentage change in uNTx within the first 4 weeks of treatment was not associated with either PFS (p value = 0.77) or OS (p = 0.26) impact. Patients with abnormal baseline uNTx (>65 nM BCE/mmol Cr) had worse outcomes, regardless of whether uNTx normalised in the first 4 weeks (data shown in supplemental appendix).
Discussion
With increasing interest in mechanisms of endocrine sensitivity and resistance pathways for MBC attempts to improve the efficacy of endocrine therapy, a number of novel strategies have been developed. These have either involved combinations of endocrine agents or combing endocrine therapy with agents that target other pathways, including Her2 [21], mTOR [22, 23], EGFR [24] and VEGF [25]. To date, the benefits of these additional therapies have been modest on the whole and not without significant toxicity.
Given that bone is the most common site of breast cancer recurrence, and the relatively long survival associated with bone predominant disease, this patient population may be an excellent one in which to explore innovative treatment strategies. VEGF, members of the VEGFR and EGFR families, are all involved in the pathogenesis of bone metastases and are clinically validated targets in breast cancer. As vandatenib, which targets VEGFR and EGFR signalling [5] and RET tyrosine kinase [6] has demonstrated modest efficacy both as a single agent [11, 12], and in combination with chemotherapy [13] in unselected patients with refractory MBC it was, therefore, hypothesised that vandatenib would result in clinical benefit when combined with fulvestrant [26] in the current proposal.
Due to the limitations in radiological response assessment in bone metastases [27], we used biomarkers of bone resorption as surrogates of response. These biomarkers have been used to assess response rates to systemic agents [15, 23, 28], optimal dose [29], frequency of administration [30], pain response [20] and choice of bone-targeted therapies [17, 31]. The primary objective of the current trial was a response (defined as a >30 % reduction from baseline at any time on treatment) of urinary N-telopeptide of type 1 collagen (uNTx). The addition of vandetanib to fulvestrant did not improve this primary outcome or any of the secondary endpoints (including PFS, OS, SREs and bone pain) compared to fulvestrant alone. Despite selecting a patient population with bone only or predominant disease, the median PFS was only 5.8 months for the combination arm and 4.8 months for the fulvestrant alone arm. These PFS values are similar to those previously observed in populations not enriched for patients with bone only or predominant disease [26].
There are a number of potential explanations for these negative results. It is possible that the dose of vandetanib, examined in this trial simply does not add to the effect of fulvestrant in terms of biomarker suppression. For medullary thyroid cancer the approved dose for single agent vandetanib is 300 mg, whereas this study used 100 mg as this was the dose chosen for combination studies with chemotherapy [9]. It is also possible that the use of uNTx, albeit the most commonly employed marker of bone resorption in clinical trials of bone-targeted agents such as bisphosphonates, is not the most appropriate marker in this setting. Interestingly, other groups have recently commented on the reduced prognostic efficacy of this marker in patients with lower NTx levels [32]. It is also possible that the study was simply not large enough to detect a difference. However, it is of note that the median PFS and OS were entirely in keeping with previous trials [26].
In an exploratory analysis, a statistically significant interaction was observed whereby baseline uNTx was predictive of an impact of FV on PFS. The prognostic importance of high baseline levels of bone turnover markers on survival has been demonstrated in bisphosphonate trials [28]. However, in the current study uNTx response was not correlated with either PFS or OS.
Study strengths included the double-blinded nature of the protocol, and the consistent processing of biochemical specimens (e.g. fasting specimens) with central laboratory analysis of biomarkers. The use of bone turnover biomarkers as a surrogate for bone metastasis response is not ideal, and is an attempt to overcome challenges in consistent radiological assessment of bone response. Indeed, as expected progression in bone was the most common reason for coming off study. This endpoint could also be affected by the lack of central radiology review. Imbalance in the randomization potentially biassing the results in favour of FV arm in terms of PFS and OS was also possible, given the higher proportion of patients having received prior palliative chemotherapy and the higher proportion of pts with non-measureable disease in the FP group. Another challenge is around the classification of patients in this study as having primary hormone resistant or secondary hormone resistant disease. This may prove particularly important as recent study results with mTOR inhibitors have suggested that certain hormone resistant behaviours may be associated with response to these agents [23].
In summary, this study showed that the addition of vandetanib to fulvestrant did not improve biomarker response, PFS or OS compared to fulvestrant alone in patients with bone-only or bone predominant disease. Further studies are required to explore the role of biomarkers as a surrogate for bone response in patients with metastatic bone disease.
References
Stopeck AT, Lipton A, Body JJ, Steger GG, Tonkin K, de Boer RH, Lichinitser M, Fujiwara Y, Yardley DA, Viniegra M, Fan M, Jiang Q, Dansey R, Jun S, Braun A (2010) Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: a randomized, double-blind study. J Clin Oncol 28:5132–5139. doi:10.1200/JCO.2010.29.7101
Santini D, Vincenzi B, Hannon RA, Brown JE, Dicuonzo G, Angeletti S, La Cesa A, Coleman RE, Tonini G, Budillon A, Caraglia M, Holen I (2006) Changes in bone resorption and vascular endothelial growth factor after a single zoledronic acid infusion in cancer patients with bone metastases from solid tumours. Oncol Rep 15:1351–1357
Amir E, Trinkaus M, Simmons CE, Dranitsaris G, Clemons MJ (2009) Vascular endothelial growth factor activity after switching of bisphosphonate treatment for metastatic breast cancer. J Clin Pathol 62:474–476. doi:10.1136/jcp.2008.062505
Poon RT, Fan ST, Wong J (2001) Clinical implications of circulating angiogenic factors in cancer patients. J Clin Oncol 19:1207–1225
Wedge SR, Ogilvie DJ, Dukes M, Kendrew J, Chester R, Jackson JS, Boffey SJ, Valentine PJ, Curwen JO, Musgrove HL, Graham GA, Hughes GD, Thomas AP, Stokes ES, Curry B, Richmond GH, Wadsworth PF, Bigley PL, Hennequin LF (2002) ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res 62:4645–4655
Carlomagno F, Vitagliano D, Guida T, Ciardiello F, Tortora G, Vecchio G, Ryan AJ, Fontanini G, Fusco A, Santoro A (2002) ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res 62:7284–7290
Plaza-Menacho I, Morandi A, Robertson D, Pancholi S, Drury S, Dowsett M, Martin LA, Isacke CM (2010) Targeting the receptor tyrosine kinase RET sensitizes breast cancer cells to tamoxifen treatment and reveals a role for RET in endocrine resistance. Oncogene 29:4648–4657. doi:10.1038/onc.2010.209
Wang C, Mayer JA, Mazumdar A, Brown PH (2012) The rearranged during transfection/papillary thyroid carcinoma tyrosine kinase is an estrogen-dependent gene required for the growth of estrogen receptor positive breast cancer cells. Breast Cancer Res Treat 133:487–500. doi:10.1007/s10549-011-1775-9
Natale RB, Thongprasert S, Greco FA, Thomas M, Tsai CM, Sunpaweravong P, Ferry D, Mulatero C, Whorf R, Thompson J, Barlesi F, Langmuir P, Gogov S, Rowbottom JA, Goss GD (2011) Phase III trial of vandetanib compared with erlotinib in patients with previously treated advanced non-small-cell lung cancer. J Clin Oncol 29:1059–1066. doi:10.1200/JCO.2010.28.5981
Wells SA, Robinson BJ, Gagel RF, Dralle H, Fagin JA, Santoro M, Baudin E, Elisei R, Jarzab B, Vasselli JR, Read J, Langmuir P, Ryan AJ, Schlumberger MJ (2012) Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol 30:134–141. doi:10.1200/JCO.2011.35.5040
Miller KD, Trigo JM, Wheeler C, Barge A, Rowbottom J, Sledge G, Baselga J (2005) A multicenter phase II trial of ZD6474, a vascular endothelial growth factor receptor-2 and epidermal growth factor receptor tyrosine kinase inhibitor, in patients with previously treated metastatic breast cancer. Clin Cancer Res 11:3369–3376
Mayer EL, Isakoff SJ, Klement G, Downing SR, Chen WY, Hannagan K, Gelman R, Winer EP, Burstein HJ (2012) Combination antiangiogenic therapy in advanced breast cancer: a phase 1 trial of vandetanib, a VEGFR inhibitor, and metronomic chemotherapy, with correlative platelet proteomics. Breast Cancer Res Treat 36:169–178
Boér K, Láng I, Llombart-Cussac A, Andreasson I, Vivanco GL, Sanders N, Pover GM, Murray E (2012) Vandetanib with docetaxel as second-line treatment for advanced breast cancer: a double-blind, placebo-controlled, randomized Phase II study. Invest New Drugs 30:681–687
Chia S, Gradishar W, Mauriac L, Bines J, Amant F, Federico M, Fein L, Romieu G, Buzdar A, Robertson JF, Brufsky A, Possinger K, Rennie P, Sapunar F, Lowe E, Piccart M (2008) Double-blind, randomized placebo controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in postmenopausal women with hormone receptor-positive, advanced breast cancer: results from EFECT. J Clin Oncol 26:1664–1670. doi:10.1200/JCO.2007.13.5822
Simmons C, Broom RJ, Cole DE, Dranitsaris G, Clemons M (2007) Urinary N-telopeptide is a rapid predictor of response to and palliative benefit from bisphosphonate therapy in patients with metastatic breast cancer. Support Cancer Ther 4:182–187
Vinholes J, Coleman R, Lacombe D, Rose C, Tubiana-Hulin M, Bastit P, Wildiers J, Michel J, Leonard R, Nortier J, Mignolet F, Ford J (1999) Assessment of bone response to systemic therapy in an EORTC trial: preliminary experience with the use of collagen cross-link excretion. European Organization for Research and Treatment of Cancer. Br J Cancer 80:221–228
Clemons MJ, Dranitsaris G, Ooi WS, Yogendran G, Sukovic T, Wong BY, Verma S, Pritchard KI, Trudeau M, Cole DE (2006) Phase II trial evaluating the palliative benefit of second-line zoledronic acid in breast cancer patients with either a skeletalrelated event or progressive bone metastases despite first-line bisphosphonate therapy. J Clin Oncol 24:4895–4900
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216
Tan G, Jensen MP, Thornby JI, Shanti BF (2004) Validation of the brief pain inventory for chronic nonmalignant pain. J Pain 5:133–137
Broom R, Du H, Clemons M, Eton D, Dranitsaris G, Simmons C, Ooi W, Cella D (2009) Switching breast cancer patients with progressive bone metastases to third generation bisphosphonates: measuring impact using the functional assessment of cancer therapy-bone pain. J Pain Symptom Manag 38:244–257. doi:10.1016/j.jpainsymman.2008.08.005
Kaufman B, Mackey JR, Clemens MR, Bapsy PP, Vaid A, Wardley A, Tjulandin S, Jahn M, Lehle M, Feyereislova A, Revil C, Jones A (2009) Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2-positive, hormone receptor-positive metastatic breast cancer: results from the randomized phase III TAnDEM study. J Clin Oncol 27:5529–5537. doi:10.1200/JCO.2008.20.6847
Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, Deleu I, Perez A, Bachelot T, Vittori L, Xu Z, Mukhopadhyay P, Lebwohl D, Hortobagyi GN (2012) Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. New Engl J Med 366:520–529. doi:10.1056/NEJMoa1109653
Gnant M, Baselga J, Rugo HS, Noguchi S, Burris HA, Piccart M, Hortobagyi GN, Eakle J, Mukai H, Iwata H, Geberth M, Hart LL, Hadji P, El-Hashimy M, Rao S, Taran T, Sahmoud T, Lebwohl D, Campone M, Pritchard KI (2013) Effect of everolimus on bone marker levels and progressive disease in bone in BOLERO-2. J Natl Cancer Inst 105:654–663. doi:10.1093/jnci/djt026
Osborne CK, Neven P, Dirix LY, Mackey JR, Robert J, Underhill C, Schiff R, Gutierrez C, Migliaccio I, Anagnostou VK, Rimm DL, Magill P, Sellers M (2011) Gefitinib or placebo in combination with tamoxifen in patients with hormone receptorpositive metastatic breast cancer: a randomized phase II study. Clin Cancer Res 17:1147–1159. doi:10.1158/1078-0432
Wagner AD, Thomssen C, Haerting J, Unverzagt S (2012) Vascular-endothelial-growth-factor (VEGF) targeting therapies for endocrine refractory or resistant metastatic breast cancer. Cochrane Database Syst Rev 7:008941
Di Leo A, Jerusalem G, Petruzelka L, Torres R, Bondarenko IN, Khasanov R, Verhoeven D, Pedrini JL, Smirnova I, Lichinitser MR, Pendergrass K, Garnett S, Lindemann JP, Sapunar F, Martin M (2010) Results of the CONFIRM phase III trial comparing fulvestrant 250 mg with fulvestrant 500 mg in postmenopausal women with estrogen receptor-positive advanced breast cancer. J Clin Oncol 28:4594–4600
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247
Lipton A, Costa L, Coleman RE (2011) Bone turnover markers: tools for prognosis and monitoring response to bisphosphonates? Breast Dis 33:59–69. doi:10.3233/BD-2010-0327
Fizazi K, Lipton A, Mariette X, Body JJ, Rahim Y, Gralow JR, Gao G, Wu L, Sohn W, Jun S (2009) Randomized phase II trial of denosumab in patients with bone metastases from prostate cancer, breast cancer, or other neoplasms after intravenous bisphosphonates. J Clin Oncol 27:1564–1571
Amir E, Freedman O, Carlsson L, Dranitsaris G, Tomlinson G, Laupacis A, Tannock IF, Clemons M (2013) Randomized feasibility study of de-escalated (Every 12 wk) versus standard (Every 3 to 4 wk) intravenous pamidronate in women with low-risk bone metastases from breast cancer. Am J Clin Oncol 36:436–442
Clemons M, Dranitsaris G, Ooi W, Cole DE (2008) A phase II trial evaluating the palliative benefit of second-line oral ibandronate in breast cancer patients with either a skeletal related event (SRE) or progressive bone metastases (BM) despite standard bisphosphonate (BP) therapy. Breast Cancer Res Treat 108:79–85
Lipton A, Cook R, Brown J, Body JJ, Smith M, Coleman R (2013) Skeletal-related events and clinical outcomes in patients with bone metastases and normal levels of osteolysis: exploratory analyses. Clin Oncol 25:217–226
Acknowledgments
Funding for this study was received from AstraZeneca. The funding source had no role in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication. We are grateful to the patients and research staff involved in this study and the members of the Data Safety Monitoring Committee: Dr. Joel Singer (University of British Columbia), Dr. Charles Geyer (CTNeT—Statewide Clinical Trials Network of Texas), Dr. Frederick R. Rickles (The George Washington University) and Dr. P. J. Devereaux (McMaster University). Preliminary results were presented at the 2013 ASCO meeting, abstract number 574. The following (and site) were co-investigators on this study; Dr. Som Mukherjee (Juravinski Cancer Centre, Hamilton), Dr. Xinni Song (The Ottawa Hospital Regional Cancer Centre), Dr. Nadia Califaretti (Grand River Regional Cancer Centre), Dr. Jose Chang (R.S. McLaughlin Durham Regional Cancer Centre), Dr. Scott Young (Health Sciences North, Sudbury), Dr. Christine Brezden-Masley St. Michael’s Hospital, Toronto), Dr. Sunil Verma (Sunnybrook Odette Cancer Centre, Toronto), Dr. David Warr (Princess Margaret Hospital, Toronto), Dr. Stephen Chia (BCCA—Vancouver), Dr. Sanraj Basi Cross Cancer Institute), Dr. Daniel Rayson (QE II HSC—Nova Scotia Cancer Centre), Dr. Florence Plaza Arnold (Saskatoon Cancer Centre), Dr. Andre Robidoux (CHUM, Montreal).
Conflict of interest
Mark Clemons: Honoraria for talks and attendances at advisory boards: AstraZeneca. Brandy Cochrane: no conflicts to declare. Gregory R Pond: no conflicts to declare. Nadia Califaretti: Honoraria for attendances at advisory boards: AstraZeneca. Stephen K. L. Chia: Honoraria for attendances at advisory boards: AstraZeneca. Rebecca Alexandra Dent: Honoraria for attendances at advisory boards: AstraZeneca. Xinni Song: no conflicts to declare. Andre Robidoux: Honoraria for attendances at advisory boards: AstraZeneca. Sameer Parpia: no conflicts to declare. David Warr: no conflicts to declare. Daniel Rayson: Honoraria for attendances at advisory boards: AstraZeneca. Kathleen I. Pritchard: Honoraria for talks and attendances at advisory boards: AstraZeneca. Mark N Levine: no conflicts to declare.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Clemons, M.J., Cochrane, B., Pond, G.R. et al. Randomised, phase II, placebo-controlled, trial of fulvestrant plus vandetanib in postmenopausal women with bone only or bone predominant, hormone-receptor-positive metastatic breast cancer (MBC): the OCOG ZAMBONEY study. Breast Cancer Res Treat 146, 153–162 (2014). https://doi.org/10.1007/s10549-014-3015-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-014-3015-6