
Abstract
Steroid sulfatase (STS) inhibition may have a therapeutic role in suppression of endocrine-responsive breast cancer. This study aimed to determine the optimal biological dose and recommended dose (RD) of the STS inhibitor irosustat. A three-part, open-label, multicenter, dose escalation study of irosustat in estrogen receptor-positive breast cancer patients involved administration of a single dose of irosustat with a 7-day observation period; followed by a daily oral dose of irosustat for 28 days; and an extension phase, in which the daily oral dose of irosustat was continued at the discretion of the investigator and as long as the patient was benefitting from the treatment. Five doses of irosustat were tested (1, 5, 20, 40, and 80 mg) in 50 patients. After 28 days of daily administration of irosustat, all the evaluated patients in the 5, 20, 40, and 80 mg cohorts achieved ≥95 % STS inhibition in peripheral blood mononuclear cells and corresponding endocrine suppression. The maximum tolerated dose was not reached, and the 40 mg dose was established as the RD. The median time to disease progression in the 40 mg cohort was 11.2 weeks. Disease stabilization was achieved in 10 % of patients potentially indicative of drug activity. Dry skin was the most frequent adverse event. The RD of irosustat is 40 mg. Disease stabilization occurred in 10 % of this heavily pretreated patient population. A larger study is required to define an accurate response rate to irosustat as a single agent and whether co-administration with an aromatase inhibitor is needed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
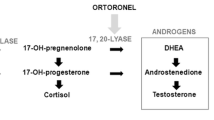
Steroid sulfatase (STS) is responsible for the hydrolysis of steroid sulfates such as estrone sulfate (E1S) and dehydroepiandrosterone sulfate (DHEAS) to estrone (E1) and dehydroepiandrosterone (DHEA). These can then be converted to estradiol (E2) and testosterone, respectively. In breast cancer, STS mRNA is increased and has prognostic significance [1–6]. Furthermore, STS is expressed in breast cancer biopsies after resistance to aromatase inhibitors (AI) [7], and is up-regulated compared to levels measured before the treatment with AI [8]. Thus, STS inhibition may have a therapeutic role in the treatment of endocrine-responsive breast cancer.
In a phase I study in postmenopausal women with metastatic estrogen receptor (ER)-positive breast cancer, a lyophilized formulation of the STS inhibitor irosustat (5 and 20 mg) inhibited STS in peripheral blood leukocytes by 98 %, resulting in a decrease of E1, E2, and androstenedione [7]. STS inhibition may therefore be an important strategy in the treatment of endocrine-responsive tumors. The purpose of this study was to determine the optimal dose of a new oral formulation of irosustat, evaluate the safety profile and anti-tumor effects, study pharmacokinetics (PK) and pharmacodynamics, and monitor tumor metabolic activity using fluorodeoxyglucose positron emission tomography (FDG PET).
Patients and methods
The study was conducted at three centers in France, one center in Belgium, and one center in the UK between April 2007 and September 2010. The trial was conducted according to the Declaration of Helsinki and International Conference on Harmonization of Good Clinical Practice. All the applicable regulatory requirements, and local independent ethics committee and institutional review board approvals were obtained before starting the trial. All the patients gave written informed consent to participate in the study.
Patient selection
Inclusion criteria were: postmenopausal women over the age of 18 years, whose disease progressed after the first-line hormonal therapy for histological or cytological confirmed ER-positive locally advanced or metastatic breast cancer; a maximum of two lines of hormonal therapy, two lines of chemotherapy (either adjuvant + first line or two metastatic lines), and a maximum of one previous course of therapy for Her2 positive breast cancer; Eastern Co-operative Oncology Group (ECOG) performance status ≤2; adequate bone marrow function defined as hemoglobin >10 g/dl, neutrophil count of >1.5 × 109/l, platelet count of >75 × 109/l; adequate hepatic function as measured by serum bilirubin <1.5 upper limit of normal (ULN), and alanine aminotransferase or aspartate aminotransferase <2.5 ULN in absence of liver metastases or <5 ULN in the presence of liver metastases; adequate renal function defined as serum creatinine <1.5 ULN or creatinine clearance of ≥60 ml/min; and life expectancy of at least 12 weeks.
Exclusion criteria included: progressive central nervous system metastases; inflammatory breast cancer; significant cardiac abnormality including pre-existing cardiac failure or prolonged QTc interval (>450 ms); the use of drugs that induce cardiac toxicity, drugs acting as carbonic anhydrase inhibitors, or vitamin K antagonists; blood pressure <100/60 mmHg; uncontrolled abnormalities of serum potassium, sodium, calcium, or magnesium levels; co-existing significant disease or systemic infections; uncontrolled diabetes; malabsorption; taking biphosphonates or investigational therapies within 4 weeks before the start of the study; previous therapy for cancer within 4 weeks (or 2 months for fulvestrant, or 4 months for trastuzumab) of entry to the study; known hypersensitivity to irosustat or drugs with a similar chemical structure; current or previous alcohol abuse; or inability to give written consent.
Study design
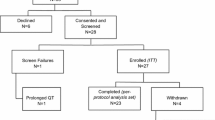
This study was a three-part open-label, multicenter, dose escalation study with once-daily administration of irosustat (Fig. 1). The primary objective was to determine the optimal biological dose (OBD) and the recommended dose (RD) of irosustat in postmenopausal women with ER-positive locally advanced or metastatic breast cancer. Secondary objectives were: to establish the safety and tolerability of irosustat; to measure STS inhibition in circulating peripheral blood mononuclear cells (PBMCs) after single and repeated doses; to measure the effect of irosustat on circulating levels of E1, E2, androstenedione, DHEA, DHEAS, E1S, androstenediol, and testosterone after single and repeated doses; to determine the PK profile of irosustat after single and repeated doses; to investigate the presence of other metabolites in addition to IDP-17619 in blood and plasma; to conduct a preliminary investigation into the clinical anti-tumor activity of irosustat and time to progression; to assess the tumor metabolic activity by FDG-PET scans; and to identify specific polymorphisms in genes involved in drug disposition and pharmacological effect.

Schematic outline of study design. DLT dose limiting toxicity, SAC safety assessment committee, MTD maximum tolerated dose
Part A involved administration of a single dose and 7 days observation. Five doses were tested in 3–6 patients: 1, 5, 20, 40, and 80 mg. Part B consisted of daily oral dosing for 28 days starting the day after the 7-day observation in Part A. The withdrawal criteria during Part B were: existence of a dose limiting toxicity (DLT) defined as National Cancer Institute Common Toxicity Criteria for Adverse Events (NCI CTCAE) grade 3 or higher drug-related toxicity [9], significant adverse events (AEs) or unacceptable toxicity, evidence of progressive disease/necessity for other cancer treatment, Fridericia’s correction of the QT interval (QTcF) >500 ms or change from baseline of >60 ms, forbidden medication, allergic reaction to the investigational product, nonadherence with protocol, or patient’s or investigator’s decision to withdraw from trial.
The OBD was defined as the dose at which all of the following were observed:
-
STS inhibition in PBMCs ≥95 % after 7 and 28 days of repeated oral daily administration;
-
maximal reduction in plasma E2 and androstenediol after 28 days of repeated oral daily administration;
-
no drug-related toxicity [grade 3 AEs (NCI CTCAE v3.0) or higher toxicity].
If no dose level met the criteria for an OBD, a RD would be defined as the dose below the maximum tolerated dose (MTD; Fig. 1).
After completion of Part B of the study, all the patients who, at the discretion of the investigator, gained clinical benefit without significant toxicity, continued to receive the same daily dose of irosustat (Part C) until withdrawal for disease progression.
Assessments
Pharmacology
E1, E1S, E2, DHEAS, androstenedione, androstenediol, DHEA, and testosterone levels were measured from plasma at baseline (before the first administration of irosustat), five times in the first 24 h, then, 120 h after the single dose (Part A), before the first of dose in Part B, after 7, 14, and 28 days of daily dosing (five times on the 28th day of daily dosing), and once monthly during Part C.
Steroid sulfatase activity was measured in PBMCs at the same time points as hormone levels except on the 28th day of daily dosing when it was measured three times.
The PK measures of irosustat and one metabolite (IDP-17619) were obtained from plasma and whole blood collected at baseline, eight times in the first 24 h, then, 72 h and 120 h after the single dose (Part A), before the first of dose in Part B, and after 7, 14, and 28 days of daily dosing (eight times on the 28th day of daily dosing).
Genotype analysis was done at baseline, 120 h after the single dose (Part A), and after 14 and 28 days of daily dosing.
Safety
All the AEs were monitored from the time that informed consent was signed until the end of the study.
Anti-tumor activity
Anti-tumor activity was explored using CT/MRI, and RECIST version 1.0 criteria and an exploratory assessment of tumor metabolic activity was assessed by 18F-FDG PET scans. Tumor assessment was performed at baseline (up to 4 weeks before first administration of irosustat), after 28 days of daily treatment, and every 6 weeks during Part C.
Laboratory analyses
DHEAS, E1S, E1, E2, androstenedione, androstenediol, DHEA, and testosterone levels were determined in plasma by high-performance liquid chromatography-MS/MS (HPLC-MS/MS). The activity of STS in blood was measured by monitoring the conversion of 3H-E1S to 3H-E1 in extracts of PBMCs. The concentrations of irosustat and IDP-17619 in plasma and whole blood were analysed using liquid chromatography and tandem mass spectrometry (SPE-LC-MS/MS).
Genotype analysis was carried out using molecular genetic techniques to amplify and analyze polymorphisms in CYP1A2, CYP2C8, CYP2C9, CYP3A5, CYP17A1, SULT1A1, SULT1A2, UGT1A3, UGT2B15, and UGT1A9. Samples were analyzed by Beckman Coulter Genomics GmbH, Benried, Germany.
Statistical analysis
Four populations of patients were used for the analysis; the safety and intention-to-treat (ITT) populations were all the patients who received at least one administration of the active treatment. The modified intention-to-treat (MITT) population consisted of patients in the ITT population who had a baseline and post-baseline assessment (7 and 28 days of repeated irosustat administration). The PK valid population was all the patients for whom plasma AUC calculations following a single dose and repeated dose of irosustat were determined. The main PK parameters of irosustat and IDP-17619 in plasma and whole blood were determined using a noncompartmental analysis (WinNonlin 5.2, Pharsight Corporation) following the different oral doses of irosustat. Analyses were performed after a single dose and once steady state was achieved.
The primary efficacy endpoint was the combined evaluation of STS inhibition in PBMCs ≥95 % after 7 and 28 days of daily administration of irosustat compared with the pretreatment values calculated as a percentage change from baseline. The reduction in plasma E2 and androstenediol levels after 28 days of daily administration of irosustat were described by cohort as a geometric mean, standard error (SE), and 95 % confidence interval (95 % CI).
Secondary efficacy parameters included the number and percentage of patients with STS inhibition in PBMCs ≥95 % at baseline and after 1, 5, 7, and 21 days post-treatment with irosustat, plasma hormonal inhibition for each hormone were described by geometric mean, SE, geometric mean ratio, and 95 % CI. Metabolic response rates of the tumors using PET scans were calculated according to RECIST criteria version 1. Time to progression was recorded as the period from the start of treatment until the criteria for disease progression was first met or until death due to tumor progression, malignant disease, or date of last news. Kaplan–Meier analyses were carried out for the 40 mg and combined (primary OBD/RD and extension) cohorts and for all the cohorts combined.
Results
Patients
Forty patients were recruited for potential inclusion in determination of the OBD (Part A). Five patients were excluded and did not receive study medication (four had QTc prolongation and one withdrew consent). Therefore, 35 patients were included for determination of the OBD, and 15 further patients were included in the extension cohort (Table 1). The median duration of exposure to irosustat was 35 days for the 1 mg dose, 56 days at 5 mg, 105 days at 20 mg, 91 days at 40 mg (primary and OBD/RD cohort), 58 days at 40 mg (extension cohort), and 91.5 days at 80 mg.
In Part A [at least a single dose of irosustat 1 mg (n = 3), 5 mg (n = 7), 20 mg (n = 6), 40 mg (n = 7), and 80 mg (n = 6)], one patient (receiving a single 5 mg dose) discontinued due to QTc prolongation. However, this patient had a history of prolongation of QTc on ECG grade 1, and the QTc interval remained prolonged at the same level during the study—the event was unlikely related to treatment. In Part B (including 21 additional patients receiving the OBD/RD 40 mg), two patients discontinued treatment: disease progression was observed after 4.9 weeks in one patient receiving irosustat 5 mg/day, and after 3 weeks in one patient receiving irosustat 40 mg/day.
Pharmacodynamics and OBD determination
After 7 days of daily administration: all the patients in the 1, 5, and 80 mg groups, 5/6 patients in the 20 mg group, and 5/7 patients in the 40 mg group achieved ≥95 % STS inhibition. After 28 days of daily administration, all the evaluated patients in the 5, 20, 40, and 80 mg groups achieved ≥95 % STS inhibition as did one of the two evaluable patients in the 1 mg group.
Geometric mean ratios of E2 levels after 28 days of daily administration of irosustat relative to baseline levels were reduced by 28–75 % in the 1, 5, 20, 40, and 80 mg groups (Fig. 2a). The 40 mg group was the only group with a significant reduction [0.72 (95 % CI 0.54–0.96)].

Geometric mean of a plasma E2 levels excluding outlier values (anomalous values that were not confirmed by reanalysis due to lack of plasma volume) and b androstenediol levels (MITT populations)
Geometric mean ratios of androstenediol after 28 days of daily administration of irosustat were reduced by 59–81 % (Fig. 2b). The reduction in plasma androstenediol was apparent 7 days after the single dose of irosustat in all the groups except the patients receiving 1 mg. Reductions in androstenediol were significant except in the 80 mg cohort. No clear dose-response relationship was evident for the reductions in plasma E2 or androstenediol levels (Fig. 2).
One patient was hospitalized for fatigue, which was considered a grade 3 drug-related serious adverse event (SAE) when receiving the 80 mg dose. This event occurred after 114 days on treatment and was not considered to be a DLT. Nevertheless, as the drug had been well-tolerated at the previous doses and the patients had a positive rechallenge at the previous dose, the 80 mg dose was assessed to be the MTD in this study. Thus, based on the full inhibition of STS activity at the 5–80 mg doses, the maximal reduction of E2 and androstenediol at all doses and the tolerability of the compound at all doses excluding 80 mg, the OBD could be 5–40 mg. The RD was determined as 40 mg because that was the dose where the smallest variation in E2 reduction was observed.
Plasma E1S levels increased from baseline in each of the five dosage groups. Geometric mean ratios of E1S levels after 28 days of daily administration of irosustat relative to baseline levels ranged from 1.21 to 3.52 pmol/l in the five dose groups. Plasma DHEA levels decreased from baseline in the 5, 20, 40, and 80 mg groups, and DHEAS levels increased in all the five dose groups. The ratio of the mean geometric ratios of DHEAS:DHEA after 28 days of daily administration of irosustat ranged from 2.00 (95 % CI 1.37–2.92) to 6.59 (95 % CI 2.15–20.20) in the five dose cohorts. No dose-response relationship was evident for changes in plasma E1, E1S, androstenedione, DHEA, DHEAS, or testosterone levels.
Pharmacokinetics
Irosustat was detected in plasma at 1 h (1 mg), 30 min (5 and 20 mg), and 10 min (40 and 80 mg) after a single dose, and remained detectable after 5 days (1 and 5 mg) or 7 days (20–80 mg). T max and T ½ after a single dose are shown in Table 2. The PK profile of irosustat in plasma after single doses and at steady state conditions is shown in Fig. 3.

Mean ± SD irosustat plasma concentration–time profiles after single and repeated oral doses of irosustat (all patients; semilogarithmic plots)
Irosustat is highly bound to erythrocytes and the mean exposure in whole blood is 166–586-fold higher than in plasma. Blood clearance is very low (around 0.11 l/h), 100-fold lower than the hepatic blood flow, and constant for doses between 1 and 20 mg. At 40 and 80 mg doses, the blood clearance increased to 0.19 l/h, indicating a tendency to non-dose proportionality in the dose range of 1–80 mg. The moderate variability in the clearance of the compound cannot be explained by the different phenotypes assayed (CYP2C9, CYP2C19, and CYP3A5). The metabolite IDP-17619 showed around 30 % of the exposure of the parent compound in plasma with a similar half-life to irosustat. In whole blood, the parent compound is the main circulating compound. In plasma, the parent compound and several glucuronides of irosustat and of IDP-17619 were circulating.
Safety
Treatment-emergent adverse events (TEAEs) were observed in all, except one patient, and thought to be treatment-related in 38 (76 %) patients (Table 3). Six patients experienced a grade 3 treatment-related AE: fatigue (two patients receiving the 40-mg dose); metastatic pain (one patient receiving the 40-mg dose); anorexia (one patient receiving the 40 mg dose); dry skin, exfoliation, and fatigue (one patient receiving the 40-mg dose); and anorexia and fatigue (one patient receiving the 80-mg dose).
There were four SAEs reported that were thought to be related to the study drug in two patients: one patient receiving the 80 mg dose of irosustat was hospitalized for fatigue (grade 3) after 114 days of treatment (the event recurred after positive re-challenge with irosustat and treatment was withdrawn); one patient receiving the 40 mg dose of irosustat reported anorexia (grade 3), nausea, and vomiting (grade 2) requiring hospitalization after 30 days of daily treatment (study medication was withdrawn before hospitalization). SAEs in two other patients were not thought to be treatment-related. There were two deaths due to disease progression during the study; neither of the two was related to the study medication.
PET results
Six patients in the OBD/RD cohort were assessed with FDG PET. Of these six patients, three patients displayed combined significant median decreases in maximum value (SUVmax) and hypermetabolic tissular volume, while three patients did not.
Pharmacogenetics
Plasma clearance of irosustat was not dependent on any of the genotypes of CYPs 1A2, 3A5, 2C8, 2C9, 2C19, 17A1; SULTs 1A1, 1A2; UGTs 1A3, 2B15. A total of 96 samples from 28 patients distributed over all the doses were analyzed. A retrospective analysis approach was applied to assess any correlation between gene expression profile and clinical outcome, and the AL035301 gene was identified with a significantly lower level of expression in 11 patients with stable disease within the duration of this study (4 months) compared with patients with progressive disease.
Time to progression
No complete or partial response was observed during the study. Five patients (10 %) remained progression-free for at least 24 weeks (33.1 weeks in one patient receiving 20 mg, 72.3, 28.4, and 27.1 weeks in three patients receiving 40 mg, and 30.7 weeks in one patient receiving 80 mg). The median time to progression was: 5.1 (range 5.1–22) weeks for patients receiving the 1 mg dose; 5.6 (range 4.9–21.7) weeks in the 5 mg group; 13.1 (range 4.9–33.1) weeks in the 20 mg group; 10.1 (3.0–72.3) weeks in the 40 mg group; and 5.0 (4.9–11.2) weeks in the 80 mg group.
Discussion
Based on these findings, we propose an OBD/RD for irosustat of 40 mg once daily; however, lower doses are also very effective at inhibiting STS in PBMCs in postmenopausal women with ER-positive breast cancer. The SAEs thought to be treatment-related were observed with the 40 and 80 mg dose levels; thus, a suitable starting dose of irosustat may be 40 mg. A reduction in dose to 20 mg could be recommended, if tolerability issues or side-effects are observed. The most frequent AE of dry skin may be readily manageable with, for example, emollient cream.
Although no objective responses were seen with irosustat according to RECIST criteria, there was evidence of clinical efficacy in this phase I study. The response rate in this heavily pre-treated group of patients is likely to be only 5–10 % with effective endocrine therapy [10], and the median time to progression of 10 and 13 weeks in the 40 and 20 mg groups, respectively, compares with a median time to progression of approximately 16 weeks in the EFECT trial of fulvestrant and exemestane [10]. Disease stabilization is often taken to be a reliable indicator of effectiveness of a novel therapy, especially endocrine therapy; the rate of 10 % having stable disease for 24 weeks or more is indicative of irosustat anti-tumor activity. A larger study will be needed to define an accurate response rate.
In this study, all the patients discontinued their AI before therapy with irosustat. In postmenopausal women, there are two pathways for estrogen formation, one relying on aromatase activity and one on STS activity. It is apparent that aromatase inhibition will fail to block the androstenediol production, and therefore, the use of STS inhibitors in combination with AI could be a potentially effective strategy to maximize the estrogen depletion. In the clinical assessment of STS inhibitors, therefore, it may be more pertinent to combine STS inhibitors with AI.
The absence of a clear dose-response in terms of effects on endocrine parameters could be due to one of several factors. One possible reason was that we did not use a sufficiently low dose. Furthermore, the most accurate measure of endocrine response to STS inhibition may be the DHEA:DHEAS ratio, which increased twofold with the doses of irosustat assessed in this study. Absence of dose-response has also been observed with AI [11, 12], which may illustrate some of the problems in measuring these endocrine parameters.
Observations are consistent with the previously published report on the metabolism of irosustat [13]; phase I and phase II liver enzymes contribute to metabolism. The metabolites were mainly monohydroxylates at the C8, C10, and C12 positions, and as here, the main metabolite was the 667-coumarin (IDP-17619). CYP2C8, CYP2C9 and CYP3A4/5 and CYP2E1 were the principal enzymes involved in the transformation of irosustat.
In conclusion, irosustat, which is well-tolerated, has PK parameters suitable for once-daily oral dosing, and can stabilize the disease in postmenopausal women with ER-positive breast cancer. The study provides proof of concept that STS is inhibited by irosustat in these patients with an effective suppression of steroid hormones in the peripheral blood. STS inhibition and treatment with irosustat is likely to be more effective when administered in combination with AI. Studies to assess this are planned.
References
James VH, McNeill JM, Lai LC, Newton CJ, Ghilchik MW, Reed MJ (1987) Aromatase activity in normal breast and breast tumor tissues: in vivo and in vitro studies. Steroids 50(1–3):269–279
Santner SJ, Feil PD, Santen RJ (1984) In situ estrogen production via the estrone sulfatase pathway in breast tumors: relative importance versus the aromatase pathway. J Clin Endocrinol Metab 59(1):29–33
Suzuki T, Nakata T, Miki Y, Kaneko C, Moriya T, Ishida T, Akinaga S, Hirakawa H, Kimura M, Sasano H (2003) Estrogen sulfotransferase and steroid sulfatase in human breast carcinoma. Cancer Res 63(11):2762–2770
Utsumi T, Yoshimura N, Takeuchi S, Ando J, Maruta M, Maeda K, Harada N (1999) Steroid sulfatase expression is an independent predictor of recurrence in human breast cancer. Cancer Res 59(2):377–381
Miyoshi Y, Ando A, Hasegawa S, Ishitobi M, Taguchi T, Tamaki Y, Noguchi S (2003) High expression of steroid sulfatase mRNA predicts poor prognosis in patients with estrogen receptor-positive breast cancer. Clin Cancer Res 9(6):2288–2293
Yoshimura N, Harada N, Bukholm I, Karesen R, Borresen-Dale AL, Kristensen VN (2004) Intratumoural mRNA expression of genes from the oestradiol metabolic pathway and clinical and histopathological parameters of breast cancer. Breast Cancer Res 6(2):R46–R55. doi:10.1186/bcr746
Stanway SJ, Purohit A, Woo LW, Sufi S, Vigushin D, Ward R, Wilson RH, Stanczyk FZ, Dobbs N, Kulinskaya E, Elliott M, Potter BV, Reed MJ, Coombes RC (2006) Phase I study of STX 64 (667 Coumate) in breast cancer patients: the first study of a steroid sulfatase inhibitor. Clin Cancer Res 12(5):1585–1592. doi:10.1158/1078-0432.CCR-05-1996
Chanplakorn N, Chanplakorn P, Suzuki T, Ono K, Chan MS, Miki Y, Saji S, Ueno T, Toi M, Sasano H (2010) Increased estrogen sulfatase (STS) and 17beta-hydroxysteroid dehydrogenase type 1(17beta-HSD1) following neoadjuvant aromatase inhibitor therapy in breast cancer patients. Breast Cancer Res Treat 120(3):639–648. doi:10.1007/s10549-010-0785-3
National Institute of Health National Cancer Institute. Common criteria for adverse events v3.0 (CTCAE) (2006). http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed 4 April 2013
Chia S, Gradishar W, Mauriac L, Bines J, Amant F, Federico M, Fein L, Romieu G, Buzdar A, Robertson JF, Brufsky A, Possinger K, Rennie P, Sapunar F, Lowe E, Piccart M (2008) Double-blind, randomized placebo controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in postmenopausal women with hormone receptor-positive, advanced breast cancer: results from EFECT. J Clin Oncol 26(10):1664–1670. doi:10.1200/JCO.2007.13.5822
Bruning PF, Bonfrer JM, Paridaens R, Nooij M, Klijn JG, Beex LV, Bruynseels J, Piccart MJ (1998) Vorozole (R83842) in the treatment of postmenopausal advanced breast cancer: relationship of serum levels of vorozole and clinical results (a study of the EORTC Breast Cancer Cooperative Group). Anticancer Drugs 9(5):419–425
Buzdar AU, Jonat W, Howell A, Jones SE, Blomqvist CP, Vogel CL, Eiermann W, Wolter JM, Steinberg M, Webster A, Lee D (1998) Anastrozole versus megestrol acetate in the treatment of postmenopausal women with advanced breast carcinoma: results of a survival update based on a combined analysis of data from two mature phase III trials Arimidex Study Group. Cancer 83(6):1142–1152. doi:10.1002/(SICI)1097-0142(19980915)83:6<1142:AID-CNCR13>3.0.CO;2-5
Ventura V, Sola J, Celma C, Peraire C, Obach R (2011) In vitro metabolism of irosustat, a novel steroid sulfatase inhibitor: interspecies comparison, metabolite identification, and metabolic enzyme identification. Drug Metab Dispos 39(7):1235–1246. doi:10.1124/dmd.111.038315
Acknowledgments
The study was funded by Ipsen. Kymos Pharma Services (Barcelona, Spain) provided hormone analysis, Beckman Coulter Genomics (formerly Epidauros Biotechnologie AG), Bernried, Germany provided pharmacogenetic analyses, Pharmacokinetics Department at Ipsen Pharma, Spain conducted PK plasma level analyses and PK/PD analyses, and the Drug Metabolism Research Laboratory at Institut Biologie et de Technologies de Saclay (LEMM; Gif-Sur-Yvette, France) did STS activity analyses. 18F-FDG-PET analysis was performed by Pr Daniel Slosman of Qualim (Genève Switzerland). Editorial support for this article was provided by Martin Gilmour of ESP Bioscience (Crowthorne, UK), funded by Ipsen. We thank the Imperial BRC and ECMC for support, as well as Cancer Research UK.
Conflict of interest
RCC, FC, NI, PSchmid and PSoulie declare that they have no conflict of interest. TL was consultant or advisor (compensated) for Roche and provided expert testimony (compensated) for Bristol-Myers Squibb within the last 3 years, and was also a consultant/advisor for GSK. CP and AK were employees of Ipsen Pharma S.A. VF was employee of Ipsen Innovation S.A. TA was an employee of Ipsen Biopharm Ltd.
Ethical standards
The trial was conducted according to the Declaration of Helsinki and International Conference on Harmonization of Good Clinical Practice. All applicable regulatory requirements, and local independent ethics committee and institutional review board approvals were obtained before starting the trial. All patients gave written informed consent to participate in the study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Coombes, R.C., Cardoso, F., Isambert, N. et al. A phase I dose escalation study to determine the optimal biological dose of irosustat, an oral steroid sulfatase inhibitor, in postmenopausal women with estrogen receptor-positive breast cancer. Breast Cancer Res Treat 140, 73–82 (2013). https://doi.org/10.1007/s10549-013-2597-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-013-2597-8