
Abstract
The purpose of this article is to determine the tumorigenic potential of estradiol treatment (E2) when combined with either progesterone (P4) or medroxyprogesterone acetate (MPA) in normal luminal human breast cells (HBE) and in human breast cancer cells (T47-D, MCF-7). Proliferation profiles were evaluated, along with the gene transactivation activity between the progesterone and glucocorticoid receptors (PR, GR) in HBE, T47-D, and MCF-7 cells treated by E2 + P4 or E2 + MPA. High throughput transcriptome analysis was performed on RNA from HBE cells treated by E2, E2 + MPA and E2 + P4. GR content was analyzed in normal breast cells as well. In HBE cells, E2 + P4 treatment was antiproliferative and promoted cellular differentiation. In contrast, E2 + MPA displayed mitogenic, antiapoptotic effects in HBE cells and did not influence cellular differentiation. The effect of P4 and MPA on cell proliferation was, however, variable in breast cancer cells. In cells containing GR or/and PR, MPA decreased proliferation whereas P4 antiproliferative effect needed the presence of PR. In HBE cells, the regulation of genes by E2 + P4, and E2 + MPA was significantly different, particularly in cell proliferation and cell death gene families. Further analysis revealed a modulation of the glucocorticoid receptor gene expression pathway by E2 + MPA. Predominant MPA glucocorticoid activity in normal and breast cancer cells was demonstrated using a glucocorticoid antagonist and the down-regulation of the GR by RNA interference. In normal luminal breast cells and in breast cancer cells, P4 and MPA combined with E2 treatment have opposing mitogenic effects due to GR. The consequences of MPA glucocorticoid potencies as well as the importance of GR in breast tissue merit a reappraisal.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hormonal replacement therapy (HRT) is commonly used to treat climacteric symptoms and prevent postmenopausal disorders. This treatment often combines estradiol (E2) or estradiol derivatives with progestogens. Progestogens were included to protect the endometrium against the development of estrogen-induced neoplasia. Today, several progestogen compounds are commercially available, and depending on the structure of these molecules, progestogens can exert additional hormonal effects in addition to their progestogenic and antiestrogenic activities. These activities are suspected to alter the risk of breast cancer. For example, the Women Health initiative randomized trial reported a small increase in breast cancer risk with HRT, including conjugated estrogens and medroxyprogesterone acetate (MPA) [1]. In a large French cohort (>80,000 women), treatment with E2 combined with progesterone (P4), or E2 combined with dydrogesterone did not increase the relative risk of breast cancer [2]. However, HRT composed of E2 in combination with different synthetic progestogens were associated with a significant increase in the breast cancer risk [2], suggesting that the use of P4 or components with pure progestogen activity, do not influence the relative risk. Presently, with the exception of France, most countries are using predominantly synthetic progestogens in HRT, occulting the comparison and analysis from studies in different countries [3].
The analysis of the proliferative activities of progestogens from published data is complicated by a number of factors. Foremost, progestogen research has occurred under different testing conditions and notably with different molecules. In addition, in a number of cases the terms progesterone and synthetic progestogens were interchangeably used despite tangible differences in their biochemistry [4].
Progesterone and synthetic progestogens have diverse steroid specificities, and consequently different pharmacological properties [5]. Studies comparing the impact of P4 and MPA combined with E2 on proliferation and differentiation in normal human breast cells and breast cancer cells are sparse. In mammary tissues collected from ovariectomized cynomolgus macaques female adults, Wood et al. [6, 7] reported that E2 combined with MPA was mitogenic and activated growth factor signalization. This effect was not seen with E2 combined with P4 treatment. The major pharmacological differences between P4 and MPA are the glucocorticoid and androgenic properties of MPA. While P4 and glucocorticoids (GC) bind to identical DNA recognition sites, the two hormones mediate very distinct biological functions [8]. Few reports have noted the importance of GC in breast cancer progression [9, 10] while in breast cancer cell lines, they were characterized for their antiproliferative and antiapoptotic activities [11, 12]. Most recently, GR activation was proposed to explain an enhanced tumor growth in rats submitted to recurrent stress [9]. However, only a few studies have reported to date on GR expression in breast cancers and no link has been reported between GR expression levels and breast cancer prognosis [13].
Breast cells are constantly exposed to estrogens, either exogenously administrated in combination with progestogens, secreted from the ovaries before menopause, or locally produced by aromatization of circulating androgens. Moreover, progestogens cellular responses and PR expression are modulated by E2 in normal and cancer breast cells. In this article, we evaluated the impact of the GC properties of MPA on normal and breast cancer cells in equivalent conditions of female hormonal plasma levels undergoing HRT. We, therefore, only compared the effects of P4 and MPA in the presence of E2. Under these conditions, we detected opposing effects of MPA and P4 on cell proliferation and gene regulation in different breast cell models. Using a glucocorticoid antagonist and GR silencing by RNA interference, we demonstrated that these effects were related to the activation of the glucocorticoid receptor of MPA in normal cells and in breast cancer cells expressing GR.
Materials and methods
Reagents and steroids
17β estradiol (E2), Progesterone (P4), medroxyprogesterone acetate (MPA), dexamethasone (DEX), insulin, Epidermal Growth Factor (EGF), transferrin, choleratoxin, cortisol, hyaluronidase, thymidine, mevinolin, ribonuclease A, were purchased from Sigma (St Quentin Fallavier, France). The progesterone receptor antagonist VA-2914 (VA) was kindly provided by HRA Pharma (Paris, France). The glucocorticoid receptor antagonist ORG-34116 (AG) was kindly provided by Dr HJ Kloosterboer ex-Organon (Oss, The Netherlands).
Cell culture procedures
T47-D and MCF-7 cell lines were, respectively, maintained in RPMI 1640 and in DMEM medium (Invitrogen, Cergy-Pontoise, France) supplemented with 10% fetal calf serum. In some experiments, cortisol (5 ng/ml) was added.
Normal human breast epithelial cell (HBE) cultures resulting from specimens of normal breast, obtained from 16 women (aged 15–25 years) undergoing surgery for reduction mammoplasty, with their informed consent according to the Ethical National rules and processed according to the French law on clinical experimentation. The patients had no reported history of breast disease and pathological studies showed only normal breast tissue. The procedure used for the HBE culture has been described elsewhere [14]. HBE cells were maintained in HAM F10 medium (Invitrogen, Cergy-Pontoise, France) containing NaHCO3 (0.24%), penicillin–streptomycin (1%), cortisol (5 ng/ml), T3 (6.5 ng/ml), choleratoxin (10 ng/ml), transferrin (5 mg/ml), insulin (0.016 U/ml) and EGF (10 ng/ml), and 5% human serum.
The steroid receptor content of the three cell types used was measured by ER-EIA (Laboratory of Hormone Biochemistry Hôpital Bicêtre). The relative amount of ER and PR between HBE and breast cancer cells was about 50–100 times different (personal data). We previously demonstrated the presence and hormonal regulation of PR and ER in our model of HBE cells [14, 15]. Nonetheless, we reconfirmed the presence of ER and PR using cytospin analysis and further characterized HBE cells as keeping a luminal differentiation using labelling positive for cytokeratine 18 (CK18) (Supplementary Fig. 1). Cytospins were then incubated with ER (6F11 Novocastra) (1:50) or PR (PGR-312 Novocastra) (1:200) (antibodies for 24 h at 4°C and a streptavidin–biotin–peroxidase method (Vectastain kit, Abcys, Paris, France).
The presence and hormonal regulation of PR and ER were verified by RT-PCR in each experiment. The Ct values corresponding to ER in HBE cells were 29.8 ± 1.6 (mean ± SEM) as compared to 25 ± 1.45 in MCF-7 cells and 21.9 ± 0.5 in T47-D cells. For PR, the Ct values were 27.0 ± 1.7, 28.7 ± 2.0 and 22.4 ± 0.9 for HBE, MCF-7 and T47-D cells, respectively (Fig. 1 Supplementary data).
Steroid treatments
After seeding, cells were starved for 24 h in serum-free and phenol red-free medium. Then cells were treated with progesterone (100 nM) or MPA (100 nM), alone or in combination with estradiol (10 nM) or with DEX (100 nM), VA (100 nM), AG (1 μM). Control cells were treated with ethanol as vehicle (1:1000). Treatments were carried out in a phenol red-free medium containing 5% dextran–charcoal-stripped serum for a period of 48 h in breast cancer cells and 96 h in HBE cells due to difference in doubling time and optimal conditions as previously reported [16–19]. Steroid concentrations were chosen according to two criteria. First, previous experiments have shown that the maximal antiproliferative effects of synthetic progestogens were observed at 100 nM [20]. Secondly, it corresponds to the plasmatic levels reported after administration of 5 mg of MPA/day (5 nM) [21]. Moreover, during the luteal phase, the level of progesterone is from 10 to 40 ng/ml.
Microarray analysis
Gene expression analysis was carried out using Illumina BeadArray Technology for Whole Genome Gene Expression (Illumina, Inc., USA). Three RNA preparations from HBE cells were used for each experimental point. The cultures were obtained from three different women. Cells were treated for 6 h with hormones then total RNA were extracted using TriZOL reagent. 10 μg aliquots of total RNA was treated with DNase I, using the “DNA free” kit (Ambion, Inc., USA) to eliminate genomic DNA contamination. Total RNA quality and quantity were assessed using the Agilent 2100 Bioanalyzer and the RNA 6000 Nano Assay kit (Agilent Technologies, USA). Only high-quality RNA, having an RNA integrity (RIN) scores above 8.0, was used for subsequent analysis.
Labelling was performed using the Illumina TotalPrep RNA Amplification kit (Ambion). Briefly, a first strand cDNA was synthesized from 500 ng of each RNA samples, than a second strand cDNA synthesis step was performed. Double-stranded cDNA was purified and biotin-labeled by in vitro transcription reaction of 12 h. The biotinylated cRNA targets were then purified and the concentration in vitro transcription products was determined using the NanoDrop 1000A Spectrophotometer (Ambion). The cRNA size distribution was evaluated by the Agilent 2100Bioanalyzer.
750 ng of amplified cRNA was hybridized to HumanHT-12 v3 Expression BeadChip arrays (Illumina, Inc., USA), according to manufacturer directions. Hybridized BeadChip arrays were stained with Amersham fluorolink streptavidin-Cy3 (GE Healthcare). BeadChip arrays were scanned with the Illumina BeadStation Scanner. The BeadStudio software v3.4.0 (Illumina, USA) was used to perform a Rank invariant normalization and to compile data values with detection scores.
Data preprocessing was performed using the R statistical package ONE CHANNEL GUY (http://www.bioconductor.org/). Filtering procedure was done employing IQR filtering function using as cut an interquantile range within the various samples lower than 0.25. Because of the small sample size, the selection step of differentially expressed genes was done using a TTest between subjects with Welch approximation and Alpha (overall threshold P value) = 0.01; the P value was based on permutation and the significance was determined by the Adjusted Bonferroni Correction (TIGR MeV, www.tigr.org).
On each list of differentially expressed genes, we performed a gene ontology (GO) analysis to assess enrichment in GO categories.
For functional pathways analysis we used a more stringent approach. Differential gene lists were crossed and only genes modulated specifically by each treatment with a fold change cut of ±1.2, were retained for successive analyses.
Statistical analyses of gene ontology (GO) terms and functional pathways analysis were performed using the web-based tool IPA (Ingenuity System, inc., http://www.ingenuity.com). This tool provides GO terms and functional pathways with their significant probabilities of enrichment (p-values from Fisher Exact test) compared with the reference gene list (Illumina HumanHT-12 v3 BeadChip arrays list). The P value accepted for significant enrichment was lower than 0.05.
Raw data and intensity values are available in GEO (record no: GSE23500).
Western blots
Cell lysates (30–80 μg) were subjected to a 10% polyacrylamide gel electrophoresis and proteins were transferred and detected using a chemoluminescence procedure. GR expression was analyzed with the human anti-GR polyclonal primary antibody (1:1000) (GR (H300), sc-8992, Santa Cruz, Heidelberg, Germany) and with an anti-rabbit HRP-conjugated secondary antibody (1:5000) (Santa Cruz, Heidelberg, Germany). Quantification of the relative expression of GR was performed using ExploraNova Morpho Expert software.
Reporter enzyme assays
Cells were transfected with MMTV-LUC and GRE-LUC plasmid reporters by using Lipofectamine Reagent or Lipofectamine LTX Reagent, respectively, in breast cell lines or HBE cells (Invitrogen, Cergy-Pontoise, France). Transfection was performed according to the manufacturer instructions. 24 h after transfection, breast cell lines and HBE cells were treated with hormones for 24 h or 48 h, respectively. At the end of the experiment, cells were lysed and luciferase activity was determined using the Luciferase Assay System (Promega, Charbonnières-les-bains, France).
GR silencing and siRNA transient transfections
MCF-7 cells and HBE cells were transiently transfected with 10 nM of either siRNA control or siRNA GR using the HiPerFect Reagent (Qiagen, Courtaboeuf, France) in MCF7 or the Lipofectamine LTX Reagent in HBE cells according to the manufacturer instructions. SiRNA GR transfection consisted in a transfection of four siRNA GR/NR3C1 sequences from Qiagen Flexitube Gene Solution GS2908 (NR3C1_5, NR3C1_6, NR3C1_1, NR3C1_2) which recognised the α-, β- and γ-GR isoforms. Cells were washed 48 h after transfection and harvested after 96 h for MCF-7 cell line and 120 h for HBE cells for experiments. For reporter transactivation experiments, cells were washed 48 h after siRNA transfection and then transfected with the GRE-LUC plasmid reporter using Lipofectamine Reagent for MCF-7 cells or Lipofectamine LTX Reagent for HBE cells. 24 h after the reporter transfection and 72 h after the siRNA transfection, cells were treated with hormones for 24 or 48 h, respectively in MCF-7 and HBE cells.
Proliferation assays
Flow cytometry analysis
Cells were synchronized with a 40 h of mevinolin at 20 μM treatment for HBE cells or with a double thymidine block for MCF-7 cells [22]. After release of synchronization, cells were washed in PBS, trypsinized and centrifuged 5 min at 1350 rpm. Cells were fixed with 70% ethanol overnight and before analysis, cells were washed in PBS and stained with 10 μg/ml propidium iodide in PBS (containing 0.835 U/ml ribonuclease A). For each sample at least 10,000 cells were counted on a BD LSR II flow cytometer (BD Biosciences, Le Pont de Claix, France). After gating out doublets and debris, cycle distribution and subG1 phase were analyzed using the ModFit LT software (Verity Software House).
Tritiated thymidine incorporation
Quantification of MCF-7 and T47-D cell proliferation was determined using thymidine incorporation under various treatments. 24 h after the beginning of hormonal treatment, cells were incubated with 2 μCi of [methyl-3H] Thymidine for 20 h for T47-D and for MCF-7 cells at 37°C. After incubation cells were washed twice with PBS 1× and once with 5% TCA (Trichloroaetic acid). Cells were incubated in 5% TCA for 15 min at 4°C and lysed in NaOH 0.1 N for 15 min at 37°C. The total cell lysate was added to 5 ml of scintillation liquid (Optiphase Hisafe 2, Perkin Elmer) and radioactivity was counted with a β counter (Beckman, LS-5000-CE).
The cell proliferation was also alternatively analysed using 5-bromo-2-deoxyuridine (BrdU) incorporation with the BrdU Cell Proliferation Assay kit (Merck, Nottingham, UK) according to the manufacturer’s protocol. Briefly, 104 cells were seeded into 96 well culture dishes and treated with hormones. 24 h after treatments, the BrdU Label (1:2000) was added to the media culture for 20 h. After fixing and denaturing, cells were successively incubated with Anti-BrdU antibody (1:100), the peroxydase goat anti-mouse IgG HRP conjugate and substrate and then absorbances were measured.
Real-time quantitative reverse transcription PCR (qRT-PCR)
Total RNA was extracted using TriZOL Reagent. 2 μg of total RNA was subjected to reverse transcription (RT) by using random primers for 1 h at 37°C. 2 μl of RT product was diluted (1:10) and subjected to quantitative PCR using sequence specific primers (300 nM) and Brillant SYBR GREEN QPCR master mix on an Mx3000P apparatus (Agilent Technologies, Massy, France). Sequence Primers for target genes were: FAS, upper, 5′-tacatcgactgcatcaggca-3′; lower, 5′-gatactttcccgtcgcatac-3′; IEX-1, upper, 5′-cggtcctgagatcttcacct-3′; lower, 5′-accctcttcagccatcagga-3′; 36B4, upper, 5′-gattggctacccaactgttg-3′; lower, 5′-caggggcagcagccacaaa-3′. Thermocycling conditions: 1 cycle at 95°C for 10 min and 40 cycles at 95°C for 30 s, 60°C for 1 min, and 72°C for 30 s. Gene expression values were normalized to the housekeeping gene 36B4.
For microarray validation, RealTime qRT-PCR were performed on cDNA corresponding to 10 ng of RNA and with the sequence-specific primers assay mix (Qiagen, Stanford, CA). Qiagen assays used in validation analysis were for: BLVRA, TMEM44, STARD13, IL6, IL33, FKBP5, FGF18, KCNN4 and IL10. Fourteen validations were done using these genes: 7 in E2 + MPA-treated cells, 5 in E2 + P4 and 2 in E2-treated cells.
Immunohistochemistry
Paraffin sections were de-waxed and rehydrated and antigens sites were retrieved by treating sections in sodium citrate buffer overnight in the Retriever 2100 apparatus (PickCell Laboratories, The Netherlands). Sections were incubated with a primary polyclonal antibody against human GR (1:75) and a streptavidin–biotin–peroxidase method (Vectastain kit, Abcys, Paris, France). A negative control (omitting the first antibody) and a positive control (normal appendix) were included in the series.
Immunofluorescence staining
Cells were grown on coverslips for 2 days. Cells were fixed successively with PFA 4% for 5 min at room temperature, with methanol and acetone at −20°C for 4 min each. Then cells were permeabilized with 1% PBS-Triton X-100 for 30 min. Cells were then incubated 2 h at 37°C with the human primary polyclonal GR (1:200) (Santa Cruz Biotechnologies) and CK18 (Sigma, St Quentin Fallavier, France) (1:800) antibodies followed by the fluorescent Cyanine 3 conjugated rabbit anti-human IgG (1:200) (Jackson ImmunoResearch, Suffolk, UK) or Alexa Fluor 488 conjugated mouse anti-human IgG (1:400) (Invitrogen, Cergy-Pontoise, France) secondary antibodies for 20 min at 37°C. Then cells were stained with DAPI and the coverslips were mounted with the Prolong Gold reagent (Invitrogen, Cergy-Pontoise, France).
Statistical analysis
To determine the statistical significance of treatments, the data were analysed using One-way ANOVA tests and multiple range Student–Newman–Keuls tests were performed to compare the relative efficiency of each treatment with the Instat 3 software (GraphPad, USA). P < 0.05 was considered as significant.
Results
Differential effect of P4 and MPA on cell proliferation
Cellular proliferation was compared in HBE, MCF-7, and T47-D cells treated with E2 alone or combined with P4 or MPA (Fig. 1a, b, c). In agreement with the known effects of E2 on breast cells, an increase of 139 ± 8% (P < 0.001), 389 ± 35% (P < 0.001), and 149 ± 19% (P < 0.001), in cell DNA content was observed in HBE, MCF-7 cells, and T47-D, respectively.

P4 and MPA on cell proliferation. Cell proliferation (a) and apoptosis (d) were measured by flow cytometry in HBE cells treated for 96 h with 10 nM E2 alone or combined with 100 nM P4 or 100 nM MPA (mean ± SEM, n = 5, *P < 0.05, **P < 0.01, ***P < 0.001). Cell proliferation was measured by [3H] thymidine incorporation in MCF-7 (b) and T47-D (c) after 48 h treatment with E2 (10 nM) alone or combined with P4 (100 nM) or MPA (100 nM) (results are expressed as % of control in mean ± SEM, n = 4 for MCF-7, n = 3 for T47-D, *P < 0.05, **P < 0.01, ***P < 0.001)
In HBE cells, P4 counteracted the E2 proliferative effects (Fig. 1a). A significant reduction in the number of cells in S and G2 phase was observed, as compared to cells treated only with E2 (P < 0.001). Consequently, a similar cellular proliferation rate occurred in cells treated with P4 in combination with E2 than in untreated cells. In contrast, MPA much less altered the E2-mediated cell proliferation (Fig. 1a). The number of apoptotic cells was also significantly increased by E2 + P4 (140 ± 10%; P < 0.001) as compared to the control whereas apoptosis was decreased in E2 + MPA treated cells (79 ± 9%; P < 0.05) (Fig. 1d).
In breast cancer cells, differential cellular growth regulation is observed. In MCF-7 cells, P4 did not counteract the effect of E2 on cell proliferation, as the proliferation rate was identical to those cells treated by E2 alone (Fig. 1b). In T47-D cells, the combined treatment of P4 with E2 resulted in a decrease of cell proliferation as compared to E2 treatment alone (22.1 ± 6.9%; P < 0.05) (Fig. 1c). In contrast to HBE cells, in MCF-7 and T47-D cells, MPA reduced the cell proliferation induced by E2 by 26.1 ± 3.6% (P < 0.01) and 15.7% ± 2.0% (P < 0.05), respectively (Fig. 1b, c). Interestingly, in MCF-7 cells, MPA was more efficient to neutralize cellular proliferation than P4. This differential effect of P4 and MPA was not seen in T47-D cells (Fig. 1c). Moreover, apoptosis analysis in MCF-7 cells demonstrated that addition of P4 or MPA did not significantly modify the apoptosis induced by E2 (data not shown). This indicates that MPA combined with E2 in MCF-7 can oppose the proliferative effects of E2, but does not act as a pro apoptotic agent in these cells.
These results suggest that P4 and MPA potencies on proliferative E2 activities are different depending if treatment is administrated to normal or breast cancer cells. In contrast, P4 and MPA induce opposite effects of apoptosis independent of the cell type.
Differential effect of P4 and MPA on cell differentiation
The differentiation state as well as the presence of factors maintaining differentiation, foretells the onset of cell transformation and neoplastic progression. If the role of P4 on mammary epithelium differentiation is well known, those of MPA are less detailed. We analyzed the expression levels of fatty acid synthetase (FAS), known to be associated with differentiation in normal breast tissue [23]. The role of FAS in breast cancer remains ambiguous since it has been associated with invasion and is negatively correlated with PR expression level [24, 25]. In HBE cells, only P4 induced FAS expression (Fig. 2a). While in MCF-7 cells, only MPA increased FAS mRNA levels (Fig. 2b), in T47-D cells both treatments P4 and MPA similarly increased FAS mRNA levels (Fig. 2c). These results suggested that in normal epithelial cells, MPA does not exhibit cellular differentiation potency. In contrast, in MCF-7, the FAS induction observed under MPA treatment could suggest a potential deleterious effect of MPA.

P4 and MPA on FAS transcription. FAS mRNA levels were determined by quantitative RT-PCR in HBE (a), MCF-7 (b), and T47-D (c). Cells were treated with E2 (10 nM) alone or combined with P4 (100 nM) or MPA (100 nM) for 48 h. Normalization was performed as indicated in “Materials and methods” section and the basal expression in untreated cells was normalized to 1 (mean ± SEM, n = 7 (MCF-7 and HBE), n = 4 (T47-D), *P < 0.05, **P < 0.01)
Differential gene regulation by P4 and MPA in normal breast cells
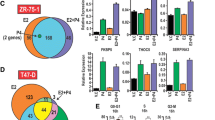
The data above described an opposite effect of MPA and P4 on cellular proliferation and differentiation in HBE cells. These observations suggest that MPA treatment could be associated with a loss of the protective effect usually conferred by P4 to normal epithelial breast cells. In order to clarify this observation, gene expression profiles of HBE cells treated with P4 and MPA in combination with E2, or by E2 alone, and compared to untreated cells was performed. As shown in Fig. 3a, only 39 genes were commonly regulated by P4 and MPA treatments, and only nine were regulated by all three treatments, suggesting that the cellular responses to these treatments are divergent. Indeed, 257 genes were regulated exclusively by E2 and P4, 204 by E2 and MPA, and 132 specific genes by E2 alone treatment (Fig. 3a).

Gene expression profiles in HBE cells treated with E2 + P4 and E2 + MPA. cDNA from three HBE cell preparations treated for 6 h with 10 nM E2 alone or combined with 100 nM P4 or 100 nM MPA were hybridized on microarrays. The gene expression profiles were analyzed and compared to untreated HBE cells. a Number of genes significantly regulated by each treatment. b Gene expression analysis sorted by major function. Statistical analyses were performed using the web-based tool DAVID Resource
We performed a GO analysis (Fig. 3b). We found significant enrichment in many categories linked to development, cellular growth and cancer in the three gene lists examined. GO analysis showed that gene families associated with cell death (P = 0.0000272) and cell growth and proliferation (P = 0.00119) were the most highly enriched by E2. After combination with P4, the profile was quite different: cell morphology and cellular function and maintenance were both highly significantly modulated (P = 0.000358 for each). In cells treated by E2 + MPA, cell death and small molecular biochemistry involved genes were the most significantly altered (Fig. 3b) (P = 0.000593 for each) in accordance with the results on apoptosis shown in Fig. 1b.
Moreover, taking into account only those genes specifically modulated by each treatment we observed significant enrichment in different pathways. In particular, E2-modulated genes were related to signalling and metabolism, taking part in RhoA signalling (P = 0.0158), lysine degradation (P = 0.0380), and glutathione metabolism (P = 0.0372). The combination of P4 with E2 mostly regulated molecules associated with metabolic functions and immune responses such as pentose phosphate pathways (P = 0.0479), galactose metabolism (P = 0.00166), folate biosynthesis (0.0138), and granzyme-A signalling (P = 0.0138). Finally, the combination of MPA with E2 regulated genes involved either in signalling or in metabolic pathways such as G-protein coupled receptor signalling (P = 0.0191), cAMP-mediated signalling (P = 0.00646), pyruvate metabolism (P = 0.0182), PXR/RXR activation (P = 0.0138), and glucocorticoid receptor signalling (P = 0.0389).
Microarray results were validated on 71% of genes (10/14) in the analysed conditions by real-time PCR (data not shown).
Effect of natural and synthetic progestogens on gene transactivation
MPA treatment modulated significantly the GR signalling pathways in HBE cells microarrays analysis. MPA has known mixed steroid properties, such as GR gene binding and transactivation, whereas P4 has no significant glucocorticoid activity [5, 26]. In order to elucidate if their opposite effects on cellular proliferation and differentiation were due to their different steroidal potencies, we further looked at the transactivation efficacy of the two progestogens on PR and GR-dependent reporter genes.
Normal and breast cancer cells were transiently transfected with the MMTV-LUC reporter vector containing both progestogen and glucocorticoid-responsive elements. In HBE and T47-D cells, P4 and MPA induced reporter transcription, (Fig. 4a, b). These effects were inhibited by the antiprogestogen, VA and the anti-glucocorticoid AG [27] (Fig. 4a, b). In contrast, we observed that only MPA-induced reporter transcription in MCF-7 cells. P4 treatment had no effect even when cells were co-treated by E2 in order to stimulate progesterone receptor (PR) expression (Fig. 4c). This latter result suggested that the induction of MPA-mediated reporter vector was not dependent on the PR cell content in MCF-7 cells. MPA MMTV-LUC reporter induction was totally counteracted by AG, and partially by VA. However, it is known that anti-progestogen also exhibits anti-glucocorticoid activities [28]. These data show that in MCF-7 cells, nuclear PR is inactive, suggesting that the MMTV-LUC reporter induction by MPA is mediated by its glucocorticoid properties.

P4 and MPA ability to activate transcription is cell type dependent. HBE (a), T47-D (b), and MCF-7 (c), cells were transfected with a murine mammary tumor virus-luciferase reporter construct (MMTV-LUC) and treated with E2 (10 nM), P4 (100 nM), MPA (100 nM), VA (100 nM), and AG (1 μM). Luciferase assays were performed 24 h after treatments. Basal luciferase activity in untreated cells was normalized to 1 for each cell type (mean ± SEM, n = 3)
GR content in normal and cancerous breast cells
GR has been reported to be present in breast cancer cell lines [13, 29, 30]. However, very little information in normal breast tissue is available [29]. We analyzed the GR content to better correlate the results obtained with MPA in our cellular models. As shown in Fig. 5a, GR expression was detected in HBE cells in the nuclei from cells expressing the luminal marker CK18. We confirmed the expression of GR in luminal epithelial and myoepithelial cells from ductal and lobular structures, as GR staining was observed in the cytoplasm and most of the nuclei when studied by immunohistochemistry in normal breast tissues from 16 patients who underwent mammoplasties (Fig. 5b). Stromal cells and adipocytes were also stained predominantly in their nuclei (not shown). Using western blot analysis, we noticed that GR was clearly expressed in normal breast cells, strongly in MCF-7 cells, but absent in T47-D cells (Fig. 5c). The ratio of GR expression was fivefold between MCF-7 cells and HBE (Fig. 5c).

Glucocorticoid receptor expression in mammary cells. Expression of GR and CK18 by immunofluorescence in HBE cells (a). Immunocytochemistry of GR labelling in red (left), CK18 in green (middle), merge (right) (×600). Immunodetection of GR in luminal (LC) and myoepithelial cells (MEC) in ductal (left) and alveolar (right) structures of normal human breast tissues (original magnification ×400). Representative images of tissues from 16 different mammoplasties (b). Detection of GR expression by western-blot in HBE, MCF-7 and T47-D cells (c). 30 μg of proteins were loaded in each well
The MPA cellular response is mediated via its glucocorticoid activity in MCF-7 cells
The GR localization studies revealed that only HBE and MCF-7 cells contained significant amount of this steroid receptor. We compared the transcription efficacy of MPA and a potent glucocorticoid, dexamethasone (DEX), on a reporter vector containing six GR-responsive elements, GRE-LUC, in normal and MCF-7 cells. MPA and DEX, but not P4, induced a strong GRE-LUC transactivation, in both HBE and MCF-7 cells (Fig. 6a, b). This promoter based activation was inhibited by AG (Fig. 6a, b). In T47-D cells, DEX and MPA had no significant effect on GRE-LUC activity (data not shown). Since T47-D cells are lacking GR (Fig. 5c), these results showed that there is no cross-activity between GR and PR for the binding and activation of GRE-LUC using a pure glucocorticoid (DEX) but also a mixed steroid (MPA) in these cells.

MPA mimics the effect of dexamethasone on transcription activity and cell proliferation. HBE (a) and MCF-7 (b) cells were transfected with a luciferase reporter construct containing glucocorticoid receptor element in the promoter (GRE-LUC) and then treated with P4 (100 nM), MPA (100 nM), DEX (100 nM), and AG (1 μM). Luciferase assays were performed 24 h after treatements (mean ± SEM, n = 3). Cell proliferation was determined by flow cytometry in HBE cells (c) and by BrdU incorporation in MCF-7 (d) cells treated with E2 (10 nM), MPA (100 nM), DEX (100 nM), and AG (1 μM) (mean ± SEM, n = 5 (HBE), n = 2 (MCF-7), *P < 0.05, **P < 0.01, ***P < 0.001)
We next examined the glucocorticoid effects on cell proliferation. In HBE cells, DEX and MPA significantly induced cell proliferation by 139 ± 5% (P < 0.001) and 123 ± 5% (P < 0.05) as compared to control (Fig. 6c). AG reversed the mitogenic effects of DEX and MPA (Fig. 6c). However, in MCF-7 cells, DEX significantly decreased cell proliferation as compared to control and E2 treated cells, by 29.2 ± 7.8% (P < 0.05) and 44.0 ± 8.6% (P < 0.001), respectively (Fig. 6d). Co-treatment of MPA and DEX with AG restored cell proliferation to the control level (P < 0.01) (Fig. 6d). These results indicated that, in HBE and MCF-7 cells, the cellular responses on cell proliferation are induced by the glucocorticoid activity of MPA.
These results strongly suggest that the difference in the P4 and MPA effects on proliferation, apoptosis and FAS expression previously observed (Figs. 1a, b, 2a, c) can be related, at least in part, to the glucocorticoid activity of MPA. In this light, we studied the MPA effects on FAS transcription. In MCF-7 cells, the level of FAS transcription induced by MPA and DEX were similar and both were inhibited by AG (Fig. 7a). To confirm the glucocorticoid-like activity of MPA in this cellular system, we studied the IEX-1 gene. This gene is implicated in cell proliferation and reported to be repressed by glucocorticoids [8]. The IEX-1 mRNA levels were decreased by MPA and DEX, and this effect was counteracted by AG (Fig. 7b).

Glucocorticoid effect of MPA on gene transcription. FAS mRNA (a) and IEX-1 mRNA (b) levels were determined by quantitative RT-PCR in MCF-7 cells treated with E2 (10 nM) alone or combined with P4 (100 nM) or MPA (100 nM) for 48 h. Normalization was performed as indicated in “Materials and methods” section and the basal expression in untreated cells was normalized to 1. Results were expressed as the mean ± SEM of at least four independent experiments
In order to confirm the participation of GR on MPA-induced cellular responses in the S phase of HBE and MCF-7 cells, GR protein content was abolished when cells were transiently transfected with GR-specific siRNA (siGR) as compared to control siRNA (siCONT) (Fig. 8a, e). In cells silenced for GR, GRE-LUC transcription activation induced by MPA was not detected (Fig. 8b, f). Similarly, DEX and MPA effects on FAS and IEX-1 mRNA levels were abolished when cell were lacking GR compared to control cells (Fig. 8c, g). We confirmed that cell proliferation induced by MPA was equally mediated by GR, since in absence of GR, MPA as DEX could not exert their proliferative or antiproliferative effects in HBE as well as MCF-7 cells (Fig. 8d, h).

GR silencing inhibited the glucocorticoid properties of MPA. HBE (a) or MCF-7 (e) cells were transfected with specific siRNA directed against GR (siGR) for 120 or 96 h, respectively. GR expression was detected by immunoblotting. Inhibition of MPA-mediated transcription by GR silencing. HBE (b) or MCF-7 (f) cells were first transfected with control siRNA (siCONT) or siGR, secondly with GRE-LUC reporter construct and then treated with MPA (100 nM), DEX (100 nM) and AG (1 μM). Luciferase assays were performed 48 or 24 h after treatments. GR silencing inhibited the gene regulation by MPA (100 nM) and DEX (100 nM) in HBE (c) or MCF-7 (g) cells. Results were expressed as the mean ± SEM of at least three and two experiments in duplicate, respectively, in MCF-7 and HBE cells. GR silencing inhibited the proliferative or antiproliferative effects of DEX and MPA, respectively, in HBE (d) and MCF-7 (h) cells. Cells were transfected with siCONT or siGR and treated with DEX (100 nM) or MPA (100 nM) for 96 or 48 h and cell proliferation was determined by flow cytometry. Results were expressed as the mean ± SEM of at least three independent experiments for MCF7, and one in triplicate for HBE
GR gene expression in breast cancer microarray studies
Glucocorticoid receptor expression regulation in breast tissue has been relatively ignored. As shown herein using breast cancer cells, their GR content may vary. We searched from the publically available information from breast gene array data bases for potential GR gene regulation within the breast tissue using Oncomine™ (Compendia Bioscience, Ann Arbor, MI). Glucocorticoid receptor was less expressed in ductal breast carcinomas than in normal breast tissues (P = 0.003, 5.31E−10 and 3.26E−6) in Sorlie et al., Perou et al.; and Richardson et al. databases, respectively [31–33]. In order to increase the number of samples, an outlier analysis was used to calculate GR gene expression variability within all genes studied in available seven datasets. GR expression was shown to be amongst the more variably expressed genes. Based on a threshold at the 5th percentile from an analysis of 731 expressed genes representing between 4,643 and 19,574 microarray experiments, the GR gene expression scored a median rank of 486 [34–40]. Globally, GR gene expression was extremely variable within breast tumors. In addition, GR gene expression variability was found within the breast carcinomas subtypes, such as in situ ductal carcinomas, invasive ductal carcinomas, and lobular carcinomas. No correlation with GR and ER or PR expression was detected, as well as with the triple negative breast carcinomas.
Discussion
The increase of breast cancer risk by combined HRT has been related to the mixed progestogen pharmacological properties [2]. The findings of this article explain some of the reasons behind this observation. Our findings demonstrate that MPA glucocorticoid properties induce opposing mitogenic, and cellular differentiation effects on normal and cancerous mammary luminal cells.
Despite an abundant literature, the role of progestogens on the breast remains controversial. Unlike most studies on progestogen effects, this study compared two combined treatments used in clinics, which differently altered the breast cancer risk, E2 + P4 and E2 + MPA. We believed that the evaluation of progestogens impact on cellular responses should be performed in presence of estradiol, because of the concomitant presence of estrogens and progesterone or progestogen in normal human breast cells. Furthermore, ER and PR can be co-expressed in breast tumors, and both exhibit biological interactions. To approach this question we used a specific cellular model of human primary luminal epithelial cells, for which the cellular responses to hormones remain similar to what is observe in normal mammary gland [14, 20, 41]. Our model shows that E2 + MPA displayed mitogenic and antiapoptotic effects in HBE cells but that P4 was able to inhibit the E2 mitogenic effect and to be proapoptotic. This result is consistent with the in vivo data, demonstrating that P4 was able to reverse the proliferative effect of E2 after topical application on breast skin [42], whereas in postmenopausal women treated with estrogen plus MPA, proliferation was increased compared to the estrogen alone in the terminal duct-lobular units of the breast [43].
Similarly, in breast tissues of Cynomolgus macaques, MPA was proliferative and stimulated the expression of EGFR ligands and its downstream targets, with a higher efficiency than P4 [6, 7]. The Cynomolgus macaque is a proper model for hormonal study response because of its close relationship on microanatomy, mammary gland development, and sexual steroid receptor expression responses to exogenous hormones compared with human [6].
These results were supported by the microarray experiments in HBE cells. We observed that E2, E2 + P4, and E2 + MPA induced different sets of genes compared to untreated cells. GO analysis showed that E2 mostly modified the gene families associated with cell death and cell growth and proliferation. Combined treatment with P4 modified the profile of genes mainly involved in cell differentiation and cell morphology, whereas E2 + MPA modified predominantly cell metabolism and cell death regulation gene sets. These data corroborated our own results on proliferation and cell death as well as the known data on cellular proliferation, differentiation, motility, and morphology regulated by estrogen and progestogens [33, 34]. The data were extracted upon hormone regulation of three individual phenotypes; the convergence of the gene regulations within the category strengthens our finding and validates our cellular model.
The modulation of the GR pathway genes by MPA is important. MPA is known to exhibit a strong relative binding affinity for GR, shown to be 45% of the DEX in ZR75-1 cells [26, 44]. We were also able to demonstrate the importance of this pharmacological aspect, as T47-D cells are lacking GR and no difference between MPA and P4 combined with E2 was observed. In the MCF-7 cells used in this study, PR gene expression was up-regulated by E2 (data not shown). However, P4 did not reverse the proliferative effects induced by E2, the MMTV reporter gene was not activated by P4 and P4 was not more quickly metabolized in a 5α-derivative as using Dutasteride, a potent 5α reductase inhibitor (data not shown). These observations suggest that PR is not functional in these MCF-7 cells indicating that the observed MPA effects should be related to its GC activity.
Our results are in agreement with the GC antiproliferative and antiapoptotic effects observed in breast cancer cell lines [11, 12]. The impact of GC on tumors remains unknown because only few reports are available on the presence of GR in breast cancers. A recent publication reported GR expression in 40% of invasive ductal carcinomas, but whereas a link between GR expression level and the age of the patients was seen, no correlation with the prognosis was observed [13]. Another publication showed that GR nuclear expression decreased with the progression of breast cancers, but GR cytoplasmic expression remained the same [30].
The contribution of GC on normal tissue is only partially documented and mostly in animal models. Glucocorticoids are involved in the proliferative phase of mice and rats mammary gland development [44]. Despite the lack of direct experimental evidence, GC are considered to induce similar effects in the human breast development. More recently, the role of GR in enhancing mammary tumor growth in rats submitted to recurrent stress was shown. Social isolation dysregulates glucocorticoid stress responses across the life span and increases glucocorticoid receptor activity in the nuclei of mammary tumor cells. This result was associated with an increase in the size, the number, and the malignancy of spontaneous mammary tumors [9].
GR expression in normal breast tissue was studied so far only in one publication [29]. In this study we observed GR labelling in the cytoplasm and the nuclei of luminal epithelial and myoepithelial cells as well as in the nuclei of stromal cells and adipocytes. Similar labeling was observed in the nuclei of ductal and lobular cells in benign breast diseases, in the stromal cell nuclei and in the nuclei of myoepithelial cells surrounding breast ducts in benign breast diseases [29]. The presence of GR in breast cancer cells irrespective of the type of breast cancer, and its widespread location to the epithelial, myoepithelial, stromal, and adipocytes as we observed in the normal tissues, suggest that the role of GC in breast cancer can be complex and unpredictable.
The role of GR expression in breast carcinogenesis has not been deeply investigated. Breast cancer microarray studies show that GR expression globally decreases between normal and tumoral tissues. But in tumoral tissues, GR expression was extremely heterogeneous and not correlated with PR or ER, as also reported in rat mammary tumors [9]. This is in accordance with the results from the clinical studies on HRT and breast cancer risk [1]. In these studies, the relative risk of breast cancer is increased to a relative low level and by a promoter effect concerning only a subset of women exposed to the treatment. The low or high expression of GR could explain that MPA independently of ER and PR could promote or repress breast tumorigenesis. This suggests that its expression level could help to define other therapeutic strategies including glucocorticoid receptor modulation.
In our study using cells containing endogenous steroid receptors, cross talk between GR and PR was not detected (Figs. 4a, c, 6a, b) suggesting that GR cannot mimic PR responses in normal and breast cancer cells. GR and PR preferentially bind specific steroids, but cross-over bindings between the specific DNA responsive elements and hormone/receptor complex have been described [45]. Despite these reported effects, PR and GR regulate different genes and encounter for very different physiological roles. How the specific action of PR and GR occurs is still not fully understood. The different physiological concentrations of P4 (10–20 ng/ml) and cortisol (100–200 ng/ml) could contribute to their different effects, as well as is the different set and sensitivity of co-modulators in their tissue specificities [46].
The limitations of this study is that the in vitro model cannot take into account the in vivo metabolism of pharmacological agents, and the interaction with the various histological components of the breast tissues. Equally, only luminal cells are contained in the culture system whereas interactions with luminal surrounded cells as stromal and myoepithelial cells might play a predominant role in GC regulation of breast tissue as these cells also express GR. Glucocorticoid effects on precancerous lesions should also be considered. More data is needed to complete the understanding of GC effects in breast carcinogenesis.
The increased risk in breast cancer under combined HRT is considered to be a promoter effect on pre-existing cancer or transformed cells [47]. We have shown that the glucocorticoid receptor is differentially expressed in breast cancer cells and its expression may modulate the MPA progestogen response. In normal breast cells, P4 is a more potent estrogen antagonist than MPA, and is a differentiation factor, whereas MPA lacks this effect. Our results suggest that MPA could promote tumorigenesis by increasing proliferation in normal cells and cancer progression in cancer cells according to PR and GR levels. MPA combined with E2 was antiproliferative, antiapoptotic, and promoted FAS expression in MCF-7 cells. The exact result of these effects is difficult to predict. This observation indicates that in healthy tissue, MPA may exert deleterious effects and could promote cancer emergence, while in cancer cells the rates of GR and PR would modulate the effects of MPA. Our results will contribute to promote complementary studies to understand the GR involvement in breast cancer development.
References
Rossouw JE, Anderson GL, Prentice RL et al (2002) Risks and benefits of estrogen plus progestogen in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA 288:321–333
Fournier A, Berrino F, Clavel-Chapelon F (2008) Unequal risks for breast cancer associated with different hormone replacement therapies: results from the E3N cohort study. Breast Cancer Res Treat 107:103–111
Bakken K, Fournier A, Lund E et al (2011) Menopausal hormone therapy and breast cancer risk: Impact of different treatments. The European prospective investigation into cancer and nutrition (EPIC). Int J Cancer 128:144–156
Calle EE, Feigelson HS, Hildebrand JS et al (2009) Postmenopausal hormone use and breast cancer associations differ by hormone regimen and histologic subtype. Cancer 115:936–945
Sitruk-Ware R (2008) Pharmacological profile of progestogens. Maturitas 61:151–157
Wood CE, Register TC, Lees CJ et al (2007) Effects of estradiol with micronized progesterone or medroxyprogesterone acetate on risk markers for breast cancer in postmenopausal monkeys. Breast Cancer Res Treat 101:125–134
Wood CE, Register TC, Cline JM (2009) Transcriptional profiles of progestogen effects in the postmenopausal breast. Breast Cancer Res Treat 114:233–242
Wan Y, Nordeen SK (2002) Overlapping but distinct gene regulation profiles by glucocorticoids and progestogens in human breast cancer cells. Mol Endocrinol 16:1204–1214
Hermes GL, Delgado B, Tretiakova M et al (2009) Social isolation dysregulates endocrine and behavioral stress while increasing malignant burden of spontaneous mammary tumors. Proc Natl Acad Sci USA 106:22393–22398
Moutsatsou P, Papavassiliou AG (2008) The glucocorticoid receptor signalling in breast cancer. J Cell Mol Med 12:145–163
TKaSR Amsterdam A (2002) Cell-specific regulation of apoptosis by glucocorticoids: implication to their anti-inflammatory action. Biochem Pharmacol 64:843–850
Lippman M, Bolan G, Huff K (1976) The effects of glucocorticoids and progesterone on hormone-responsive human breast cancer in long-term tissue culture. Cancer Res 36:4602–4609
Belova L, Delgado B, Kocherginsky M, Melhem A, Olopade OI, Conzen SD (2009) Glucocorticoid receptor expression in breast cancer associates with older patient age. Breast Cancer Res Treat 116:441–447
Malet C, Gompel A, Yaneva H et al (1991) Estradiol and progesterone receptors in cultured normal human breast epithelial cells and fibroblasts: immunocytochemical studies. J Clin Endocrinol Metab 73:8–17
Malet C, Gompel A, Spritzer P et al (1988) Tamoxifen and hydroxytamoxifen isomers versus estradiol effects on normal human breast cells in culture. Cancer Res 48:7193–7199
Cavailles V, Gompel A, Portois MC et al (2002) Comparative activity of pulsed or continuous estradiol exposure on gene expression and proliferation of normal and tumoral human breast cells. J Mol Endocrinol 28:165–175
Rochefort H (1995) Oestrogen- and anti-oestrogen-regulated genes in human breast cancer. Ciba Found Symp 191:254–265 (discussion 265–258)
May FE, Johnson MD, Wiseman LR et al (1989) Regulation of progesterone receptor mRNA by oestradiol and antioestrogens in breast cancer cell lines. J Steroid Biochem 33:1035–1041
Westley B, May FE, Brown AM et al (1984) Effects of antiestrogens on the estrogen-regulated pS2 RNA and the 52- and 160-kilodalton proteins in MCF7 cells and two tamoxifen-resistant sublines. J Biol Chem 259:10030–10035
Gompel A, Malet C, Spritzer P et al (1986) Progestogen effect on cell proliferation and 17 beta-hydroxysteroid dehydrogenase activity in normal human breast cells in culture. Journal Clin Endocrinol Metab 63:1174–1180
Svensson LO, Johnson SH, Olsson SE (1994) Plasma concentrations of medroxyprogesterone acetate, estradiol and estrone following oral administration of Klimaxil, Trisequence/Provera and Divina. A randomized, single-blind, triple cross-over bioavailability study in menopausal women. Maturitas 18:229–238
Somai S, Chaouat M, Jacob D et al (2003) Antiestrogens are pro-apoptotic in normal human breast epithelial cells. Int J Cancer 105:607–612
Chalbos D, Joyeux C, Galtier F et al (1992) Progestogen-induced fatty acid synthetase in human mammary tumors: from molecular to clinical studies. J Steroid Biochem Mol Biol 43:223–228
Kuhajda FP (2006) Fatty acid synthase and cancer: new application of an old pathway. Cancer Res 66:5977–5980
Esslimani-Sahla M, Thezenas S, Simony-Lafontaine J, Kramar A, Lavaill R, Chalbos D, Rochefort H (2007) Increased expression of fatty acid synthase and progesterone receptor in early steps of human mammary carcinogenesis. Int J Cancer 120:224–229
Poulin R, Baker D, Poirier D et al (1991) Multiple actions of synthetic ‘progestogens’ on the growth of ZR-75-1 human breast cancer cells: an in vitro model for the simultaneous assay of androgen, progestogen, estrogen, and glucocorticoid agonistic and antagonistic activities of steroids. Breast Cancer Res Treat 17:197–210
Karst H, de Kloet ER, Joels M (1997) Effect of ORG 34116, a corticosteroid receptor antagonist, on hippocampal Ca2+ currents. Eur J Pharmacol 339:17–26
Attardi BJ, Burgenson J, Hild SA et al (2004) In vitro antiprogestational/antiglucocorticoid activity and progestogen and glucocorticoid receptor binding of the putative metabolites and synthetic derivatives of CDB-2914, CDB-4124, and mifepristone. J Steroid Biochem Mol Biol 88:277–288
Lien HC, Lu YS, Cheng AL, Chang WC, Jeng YM, Kuo YH, Huang CS, Chang KJ, Yao YT (2006) Differential expression of glucocorticoid receptor in human breast tissues and related neoplasms. J Pathol 209:317–327
Conde IP, FraileB Lucio J, Arenas MI (2008) Glucocorticoid receptor changes its cellular location with breast cancer development. Histol Histopathol 23:77–85
Sorlie T, Perou CM, Tibshirani R et al (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 98:10869–10874
Perou CM, Sorlie T, Eisen MB et al (2000) Molecular portraits of human breast tumours. Nature 406:747–752
Richardson AL, Wang ZC, De Nicolo A et al (2006) X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell 9:121–132
Boersma BJ, Reimers M, Yi M et al (2008) A stromal gene signature associated with inflammatory breast cancer. Int J Cancer 122:1324–1332
Bonnefoi H, Potti A, Delorenzi M et al (2007) Validation of gene signatures that predict the response of breast cancer to neoadjuvant chemotherapy: a substudy of the EORTC 10994/BIG 00-01 clinical trial. Lancet Oncol 8:1071–1078
Chin K, DeVries S, Fridlyand J et al (2006) Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 10:529–541
Loi S, Haibe-Kains B, Desmedt C et al (2007) Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J Clin Oncol 25:1239–1246
Loi S, Haibe-Kains B, Desmedt C et al (2008) Predicting prognosis using molecular profiling in estrogen receptor-positive breast cancer treated with tamoxifen. BMC Genomics 9:239
Sotiriou C, Neo SY, McShane LM et al (2003) Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci USA 100:10393–10398
Yu K, Ganesan K, Miller LD et al (2006) A modular analysis of breast cancer reveals a novel low-grade molecular signature in estrogen receptor-positive tumors. Clin Cancer Res 12:3288–3296
Gompel A, Somaï S, Chaouat M et al (2000) Hormonal regulation of apoptosis in breast cells and tissues. Steroids 65:593–598
Foidart JM, Colin C, Denoo X et al (1998) Estradiol and progesterone regulate the proliferation of human breast epithelial cells. Fertil Steril 69:963–969
Hofseth LJ, Raafat AM, Osuch JR et al (1999) Hormone replacement therapy with estrogen or estrogen plus medroxyprogesterone acetate is associated with increased epithelial proliferation in the normal postmenopausal breast. J Clin Endocrinol Metab 84:4559–4565
Wintermantel TM, Bock D, Fleig V, Greiner EF, Schutz G (2005) The epithelial glucocorticoid receptor is required for the normal timing of cell proliferation during mammary lobuloalveolar development but is dispensable for milk production. Mol Endocrinol 19:340–349
Leo JC, Guo C, Woon CT et al (2004) Glucocorticoid and mineralocorticoid cross-talk with progesterone receptor to induce focal adhesion and growth inhibition in breast cancer cells. Endocrinology 145:1314–1321
Szapary D, Song LN, He Y et al (2008) Differential modulation of glucocorticoid and progesterone receptor transactivation. Mol Cell Endocrinol 283:114–126
Foidart JM, Desreux J, Pintiaux A et al (2007) Hormone therapy and breast cancer risk. Climacteric 10(Suppl 2):54–61
Acknowledgments
We thank Drs. Michèle Resche-Rigon (HRA Pharma, Paris) for helpful discussions and providing the anti-progestogen VA-2914, Steve K. Nordeen (University of Colorado, Denver) for kindly providing sequences of IEX-1 primers, Helenius J. Kloosterboer (Organon, Part of Shering-Plough, Oss) for providing antiglucocorticoid ORG-34116, Ramiro Castro Urology Medical Director (GSK, UK) for the generous gift of Dutasteride and Sylvie Dumont for her excellent technical assistance in immunohistochemistry experiments. We are grateful to Niel Insdorf for his kind help in editing the manuscript. This research was supported by grants from INSERM-UPMC, the Association pour la Recherche sur le Cancer, HRA Pharma and Novartis (France). Aurélie Courtin was a recipient of a grant from the Association pour la Recherche sur le Cancer, Laudine Communal is a recipient for a grant from HRA Pharma. Myriam Vilasco is a recipient of a post-doc fellowship (Institut National Contre le Cancer-INCA). Daniela Taverna was supported by grants from the University of Torino (Local Research Funding 2007/DT, 2008/DT), Regione Piemonte Ricerca Scientifica Applicata (CIPE2004/DT) and Compagnia di San Paolo, Torino. Daniela Cimino is a fellow of the Regione Piemonte.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10549_2011_1394_MOESM1_ESM.ppt
Supplementary Figure 1: Validation of HBE cell model: PR and ER protein expression. Immunocytochemistry of PR (left) and ER (right) and negative control (NC) (a) (original magnification ×200). Characterization of luminal epithelial cells using expression of CK18 detected by immunofluorescence (b) (original magnification ×600). Comparison of PR and ER mRNA amounts in normal and breast cancer cells by RT-qPCR (c). Results are expressed in cycle threshold (Ct) values (mean ± SEM, n = 5 (HBE), n = 3 (MCF-7 and T47-D))
Rights and permissions
About this article
Cite this article
Courtin, A., Communal, L., Vilasco, M. et al. Glucocorticoid receptor activity discriminates between progesterone and medroxyprogesterone acetate effects in breast cells. Breast Cancer Res Treat 131, 49–63 (2012). https://doi.org/10.1007/s10549-011-1394-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-011-1394-5