
Abstract
The c.156_157insAlu BRCA2 mutation has so far only been reported in hereditary breast/ovarian cancer (HBOC) families of Portuguese origin. Since this mutation is not detectable using the commonly used screening methodologies and must be specifically sought, we screened for this rearrangement in a total of 5,443 suspected HBOC families from several countries. Whereas the c.156_157insAlu BRCA2 mutation was detected in 11 of 149 suspected HBOC families from Portugal, representing 37.9% of all deleterious mutations, in other countries it was detected only in one proband living in France and in four individuals requesting predictive testing living in France and in the USA, all being Portuguese immigrants. After performing an extensive haplotype study in carrier families, we estimate that this founder mutation occurred 558 ± 215 years ago. We further demonstrate significant quantitative differences regarding the production of the BRCA2 full length RNA and the transcript lacking exon 3 in c.156_157insAlu BRCA2 mutation carriers and in controls. The cumulative incidence of breast cancer in carriers did not differ from that of other BRCA2 and BRCA1 pathogenic mutations. We recommend that all suspected HBOC families from Portugal or with Portuguese ancestry are specifically tested for this rearrangement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The pattern of BRCA1 and BRCA2 mutations in hereditary breast/ovarian cancer (HBOC) families varies widely among different populations. Many present a wide spectrum of different mutations throughout these genes, while some ethnic groups show a high frequency of particular mutations due to founder effects [1, 2]. Identification of founder mutations makes it possible to use more specific approaches to molecular testing [3], allowing the analysis of more patients with less stringent selection criteria in a given population. Furthermore, a frequent founder mutation in a population allows a more accurate estimation of mutation-specific cumulative cancer incidence, facilitating also identification of genetic and environmental risk modifiers.
The c.156_157insAlu BRCA2 mutation was first described by Teugels et al. [4] in a Portuguese patient residing in Belgium. These authors demonstrated that this exon 3 Alu insertion causes an in-frame deletion of that exon at the mRNA level, and thereby deletes a transcriptional activation domain [4]. Machado et al. [5] later described a regional founder effect for this rearrangement in HBOC families mostly originated from central/southern Portugal. We recently evaluated the contribution of the c.156_157insAlu BRCA2 mutation to inherited predisposition to breast/ovarian cancer in families originated mostly from northern/central Portugal [6] and found that this rearrangement is responsible for more than half of all deleterious BRCA2 mutations and about one-fourth of all deleterious mutations in HBOC families. Additionally, in light of some doubts raised about the pathogenic effect of BRCA2 exon 3 skipping [7], we demonstrated that the BRCA2 full length transcript is produced exclusively from the wild type allele in patients carrying the c.156_157insAlu BRCA2 rearrangement and that the mutant allele co-segregates with the disease in HBOC families and is absent in healthy blood donors, although minimal exon 3 skipping in BRCA2 mRNA can be found in negative controls [6, 8].
Although all reported c.156_157insAlu BRCA2 mutations have so far been identified in Portuguese HBOC families [4–6], this mutation is not detected using the common screening methodologies and must be specifically sought [4, 8], so one cannot currently rule out its presence in other populations. To gain insight into the ancestral origin and population spread of the c.156_157insAlu BRCA2 mutation, we screened for this rearrangement in 5,443 suspected HBOC patients from several countries and performed an extensive haplotype study using closely linked microsatellite markers and single nucleotide polymorphisms (SNPs) in carrier families. In addition to estimating the age of the c.156_157insAlu BRCA2 mutation, we used real-time RT-PCR to quantify the production of the transcript lacking exon 3 in carriers and non-carriers.
Materials and methods
Families
This study comprised a total of 5,443 suspected HBOC families from 13 countries in Europe, North and South America and Asia. From Portugal, 149 new suspected HBOC families were selected for BRCA1 and BRCA2 mutation screening using previously described criteria [6, 9] after written informed consent. Molecular testing at the Department of Genetics of the Portuguese Oncology Institute, Porto, Portugal (IPO-Porto) started by looking for the c.156_157insAlu BRCA2 mutation, followed by full BRCA1 and BRCA2 mutation screening with the previously reported methodology [6, 8, 9]. Additionally, screening for the c.156_157insAlu BRCA2 mutation was performed in 5,294 suspected HBOC families living in countries other than Portugal in whom no deleterious BRCA1/BRCA2 mutations had previously been found, with the following distribution: 1,209 from Spain (356 from L’Hospitalet de Llobregat, 341 from Madrid, 151 from Valladolid, 132 from Zaragoza, 123 from Santiago de Compostela, and 106 from Barcelona), 1,087 from France (650 from Clermont-Ferrand, 428 from Saint-Cloud, and nine from Villejuif, all the latter with Portuguese ancestry), 820 from Holland (Groningen), 758 from Denmark (Funen and Jutland), 400 from Greece (Athens), 219 from Switzerland (Geneva), 200 from Belgium (Brussels), 185 from Israel (Tel Aviv), 144 from Brazil (98 from Porto Alegre and 46 from S. Paulo), 103 from Canada (Montreal), 91 from India (Chennai), 75 from Italy (Rome), and three from USA (Seattle, all with Portuguese ancestry). Besides the suspected HBOC families, two consecutive series of breast cancer patients from Rio de Janeiro, Brazil (390), and Azorean Island of São Miguel, Portugal (86), were also screened for the c.156_157insAlu BRCA2 mutation. Additionally, predictive testing was performed in four individuals from two additional families (two relatives from each family living in Rhode Island, USA, and in Villejuif, France, respectively) with the c.156_157insAlu BRCA2 mutation identified elsewhere. IRB approval was obtained at each participating institution.
For the purpose of haplotype studies and age estimation of the c.156_157insAlu BRCA2 mutation, the 14 HBOC families we previously reported [6] and the family (four c.156_157insAlu carriers) initially identified by Teugels et al. [4] were also included. The geographic origin of the c.156_157insAlu BRCA2 positive families was inferred from the birthplace of the oldest carrier or of the oldest family member most likely to be a carrier.
Screening for the c.156_157insAlu BRCA2 mutation
The screening for the c.156_157insAlu BRCA2 mutation in the suspected HBOC families from Portugal, and of samples originating from the Athens, Barcelona, Madrid and Zaragoza labs, as well as the predictive testing of four individuals from two additional families living in Rhode Island and Villejuif, respectively, was performed at the Department of Genetics of IPO-Porto. The remaining cases were analyzed at the respective labs (except the cases from Rio de Janeiro, which were analyzed in Toronto) using the same protocol and a positive control provided by the Portuguese lab.
Screening for the c.156_157insAlu BRCA2 mutation was performed using two independent PCRs [6, 8], one for exon 3 amplification and another specific for the Alu rearrangement. Using this strategy, we expect two amplicons in positive cases in the first PCR (one amplicon if negative) and one amplicon in the second PCR (none if negative). The second PCR helps to control the first PCR for potential problems with preferential amplification of the shorter fragment (wild type), whereas the first PCR controls for potential absence of amplification in the second PCR. This strategy of two independent PCRs, followed by sequencing of the genomic fragments in positive cases, allows the unambiguous detection of the c.156_157insAlu BRCA2 mutation [6, 8]. Positive and negative controls were used in all experiments and all positive cases were confirmed in a second independent sample.
Real-time RT-PCR analysis
Primers and probes for the transcripts BRCA2 wild type (BRCA2-wt) and BRCA2 lacking exon 3 (BRCA2-Δex3) were designed with Primer Express 2.0 (Applied Biosystems, Foster City, USA) (Supplementary file). To determine the relative expression levels of the target transcripts in each sample, the comparative C T method was performed as described by Schmittgen and Livak [10]. The relative expression of the transcripts in two different groups (that included 10 carriers and eight controls) was calculated using the \( 2^{{ - \Updelta C_{\text{T}} }} \) method. The ratio \( 2^{{ - \Updelta C_{\text{T}} }} \) BRCA2-Δex3/\( 2^{{ - \Updelta C_{\text{T}} }} \) BRCA2-wt was calculated for each sample. The Mann–Whitney U Test was used to compare the relative expression of those transcripts between the two groups. Statistical analysis was performed with SPSS version 11 and statistical significance was considered whenever P < 0.05.
Mutation-specific cumulative incidence of breast cancer
The cumulative incidence of breast cancer in women with the c.156_157insAlu BRCA2 mutation was derived using the method of Kaplan and Meier, with unaffected individuals censored at the age of last follow-up or death without breast cancer. Only individuals shown to be carriers or obligate carriers were used for this calculation.
Microsatellite and SNP typing
Haplotype analysis was carried out in families in which the c.156_157insAlu BRCA2 mutation was detected in at least one family member in addition to the proband. A total of 15 probands and 62 family members, including the three informative families previously reported [6] and the one described by Teugels et al. [4], were genotyped for polymorphic microsatellite markers flanking BRCA2 as described [6]. The physical distances of the genetic markers were derived from the National Center for Biotechnology Information (NCBI) Map Viewer (genome build 36.3) (http://www.ncbi.nlm.nih.gov/projects/mapview/). All nine markers were assayed by PCR using fluorescently 5′-labeled primers. PCR products were run on an ABI PRISM 310 Genetic Analyser (Applied Biosystems) together with the fluorescence labeled DNA fragment size standard TAMRA.
Single-nucleotide polymorphism (SNP) markers were used to obtain a haplotype spanning ~1.1 Mb encompassing the region between the D13S260 and D13S1695 microsatellite markers, where the first recombinant and/or mutational events were observed. In order to capture most of the genetic variation in this region and to avoid redundant SNP markers (i.e., markers in strong linkage disequilibrium), we performed Tag-SNP, namely Tagger Multimarker, using International HapMap Project CEPH (Utah residents with ancestry from northern and western Europe) population data (www.hapmap.org). We developed SNaPshot assays for 19 SNP markers by multiplexed nucleotide primer extension reaction using dye label terminators (Applied Biosystems). The primers for multiplex amplification and single base extension (Supplementary file) were designed using the online Primer-BLAST tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). AutoDimer (www.cstl.nist.gov/strbase/NIJ/AutoDimer.htm) was used to test for potential hairpin structures and primer dimers. The 19 SNPs were PCR amplified in four multiplex reactions with amplicon length between 100 and 450 bp. The multiplex SNaPshot reaction and capillary electrophoresis was done following the manufacturer’s protocol (Applied Biosystems).
Haplotype construction and estimation of mutation age
Haplotype construction was performed manually based on the genotypes obtained of index cases and family members. We estimated the age of the c.156_157insAlu BRCA2 mutation from the variation accumulated in their ancestral haplotypes, as described by Martins et al.[11] This method takes into account both recombination (c) and mutation (μ) rates in the generation of variation. The probability of change per generation (ε) is given by ε = 1−[(1 − c)(1 − μ)], and the average of mutation and recombination events (λ) equals εt, where t is the number of generations. The recombination rate (c) was estimated from the physical distance between the two most distant markers (D13S1700 and D13S267) using a conversion factor calculated in Rutgers Map Interpolator (http://compgen.rutgers.edu/old/map-interpolator/). The estimate of average mutation rate used was 7.8 × 10−4 [12] for dinucleotides and two times lower for tetranucleotides.
Results
Detection of the c.156_157insAlu BRCA2 mutation
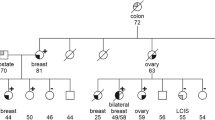
Of the 149 Portuguese probands studied for germline mutations in the BRCA1 and BRCA2 genes at IPO-Porto, 11 patients presented the c.156_157insAlu BRCA2 mutation (Fig. 1) and 18 patients presented other deleterious mutations in either BRCA1 (10 patients) or BRCA2 (8 patients) genes (data not shown). Together with the 14 probands we previously reported with this mutation [6], a total number of 25 HBOC families with the c.156_157insAlu BRCA2 rearrangement had been identified at IPO-Porto at the time of writing. Altogether, 68 individuals from these 25 HBOC families have so far been tested for the c.156_157insAlu BRCA2 mutation and 39 of them were shown to be carriers of the mutant allele. The geographic origins of all the c.156_157insAlu BRCA2 positive families are shown in Supplementary Fig. 1. Although most of the families originated from northern/central Portugal, most likely reflecting our target population for genetic testing, we also detected the c.156_157insAlu mutation in families from southern Portugal and Madeira Island.

Molecular diagnosis of the BRCA2 c.156_157insAlu mutation using two independent PCR analyses, showing positive cases in lanes 1 and 2 and a negative case in lane 3. Lane 4 corresponds to a positive control and NTC is a non template control. MW refers to 100 bp DNA standard. (a) PCR specific for BRCA2 exon 3, showing an additional band resulting from the insertion of a DNA fragment of about 350 bp long within exon 3 of BRCA2 in positive cases. (b) PCR specific for the c.156_157insAlu BRCA2 mutation, showing an amplicon in positive cases. (c) Sequence electrophorograms of the amplified genomic fragment of a mutation positive case (forward, top; reverse, bottom), confirming the Alu insertion (arrow) in BRCA2 gene exon 3. The Alu insertion is flanked by a short sequence duplication (TSD) as previously described by Teugels et al. [4]
Of the 5,294 suspected HBOC families with no known deleterious mutation originating from other countries, only one proband tested in Clermont-Ferrand was shown to carry the c.156_157insAlu BRCA2 mutation. Interestingly, this patient belongs to a family of Portuguese origin living in France. Additionally, the two relatives living in Rhode Island (family with origin in Mangualde, central Portugal) and the two relatives living in Villejuif (family with origin in Porto, Portugal) for whom we performed predictive testing, were carriers of the c.156_157insAlu BRCA2 mutation that had previously been identified elsewhere in Portuguese family members. Finally, the patient originally reported by Teugels et al. [4] belongs to a Portuguese family originally from the region of Guarda (central Portugal).
Quantitative transcript analysis
Real-time RT-PCR showed quantitative differences between the full length and the BRCA2-Δex3 transcripts in c.156_157insAlu BRCA2 mutation carriers and controls. The relative expression of the BRCA2-Δex3 transcript was sixfold higher in carriers compared with controls, whereas a threefold decrease was observed for the BRCA2-wt transcript in patients compared with controls (Fig. 2). The difference observed between patients and controls was statistically significant (P = 0.00032).

Real-time RT–PCR quantification of the altered transcript ratios in BRCA2 c.156_157insAlu carriers as compared with controls. The relative expression of the BRCA2-Δex3 transcript was sixfold higher in carriers compared with controls, whereas a threefold decrease was observed for the BRCA2-wt transcript in patients compared with controls
Mutation-specific cumulative incidence
Using the method of Kaplan and Meier, the cumulative incidence of breast cancer in women carrying the c.156_157insAlu BRCA2 mutation was 90% until the age of 60 years (Fig. 3).

Cumulative incidence of breast cancer among c.156_157insAlu BRCA2 germline mutation carriers, reaching 90% at about 60 years of age
Ancestral STR-based haplotypes and age estimate
Nine different haplotypes were phased for 11 out of the 15 families, three of them reported earlier [6]. The results of the haplotype analyses for the 11 informative families are shown in Table 1 and the most parsimonious relationships among flanking haplotypes are presented as a phylogenetic network in Fig. 4. The probability of mutation versus recombination was evaluated, considering the minimum number of stepwise mutations. In the 11 informative families, SNP haplotypes were constructed in order to establish if a specific microsatellite was different from the consensus because of a recombination event rather than a mutation (Supplementary Fig. 2).

Phylogenetic network showing the most parsimonious relationships among flanking short tandem repeat-based haplotypes in families carrying the c.156_157insAlu BRCA2 mutation. Circle and line sizes are proportional to the number of families and stepwise mutations, respectively. Diamonds indicate recombination events. When it was not possible to determine if the most parsimonious relationship was due to a stepwise mutation or a recombination event we represented it by dashed diamonds
Based on the mutation and recombination events observed in microsatellite haplotypes and assuming a generation time of 25 years, the age estimate for the c.156_157insAlu BRCA2 mutation is 558 ± 215 years (Table 1).
Discussion
The c.156_157insAlu BRCA2 mutation has so far only been reported in HBOC families of Portuguese origin. Here we show that this rearrangement accounts for 57.8% of the BRCA2 mutations and 37.9% of all deleterious mutations in HBOC families originating mostly from northern/central Portugal. This study confirms our and other earlier findings indicating that this is by far the most common BRCA mutation in Portuguese families with hereditary predisposition to breast/ovarian cancer, being detected in about 8% of all probands tested and presenting a nation-wide distribution [5, 6]. This high frequency makes it cost-effective to test specifically for this rearrangement prior to screening the entire coding regions of BRCA1 and BRCA2 in suspected HBOC families from Portugal or with Portuguese ancestry. Furthermore, complementing earlier data showing that the c.156_157insAlu BRCA2 mutation leads to skipping of exon 3 [4] and that minimal exon 3 skipping in BRCA2 mRNA can be found in negative controls [6, 8], we here demonstrate by real-time RT-PCR that carriers present significantly more BRCA2-Δex3 transcripts and much less full length transcripts than controls. We further show that the cumulative incidence of breast cancer in c.156_157insAlu BRCA2 mutation carriers does not differ from that of other BRCA2 and BRCA1 pathogenic mutations in our population (data not shown) or elsewhere [13], further strengthening its role as the major contributor to hereditary predisposition to breast cancer in Portugal. Although the function, if any, of the BRCA2 exon 3 skipping seen in controls is unknown, the observed penetrance in c.156_157insAlu carriers would not be expected if this transcript was fully functional. We therefore conclude that this BRCA2 rearrangement causes hereditary breast/ovarian cancer because the mutated allele is only able to give rise to BRCA2-Δex3 transcripts and not to BRCA2-wt transcripts.
Since the c.156_157insAlu BRCA2 mutation had only been reported in HBOC families of Portuguese origin [4–6] and is not detectable with commonly used screening methodologies, one can not exclude that it is present in other populations until it is specifically sought. To further evaluate whether or not it constitutes a population-specific founder mutation, we screened for the c.156_157insAlu BRCA2 rearrangement outside Portugal in more 5,294 suspected HBOC families with no known deleterious BRCA1/BRCA2 mutations coming from several countries mainly from Europe, but also from Asia and North and South America. In addition to the family identified in Belgium by Teugels et al. [4], we now detected this mutation in one proband living in France and in four individuals requesting predictive testing living in France and in the USA, all having in common the fact that they are relatively recent immigrants of Portuguese origin in those countries. Interestingly, c.156_157insAlu BRCA2 mutation was not detected in 1,209 suspected HBOC families from Spain, including those from Galicia, the Spanish region with which Portugal shares more linguistic and cultural links, as also demonstrated by our recent finding of a common ancestry for the Portuguese HBOC families presenting the R71G BRCA1 founder mutation of Galician origin [14].
Our findings indicate that, within the relatively large sample population studied, the c.156_157insAlu BRCA2 mutation is unique to HBOC families of Portuguese ancestry, a fact that is hardly compatible with the age of about 2500 years previously estimated by Machado et al. [5]. Although geographic distribution of mutations is only an indirect measure of mutation age, more widespread mutations tend to be older than mutations showing a regional distribution, with the development of urbanization and industrialization in the past 700 years leading to rapid populations growth and therefore to the recent appearance of vast numbers of new alleles, some of which cause hereditary breast/ovarian cancer, each being specific to one population or even to one family [15]. In order to get a more accurate mutation age estimate of the c.156_157insAlu BRCA2 rearrangement, we performed an extensive haplotype analysis having in mind that the size of an ancestral haplotype around a mutation is inversely correlated with the number of generations separating the common ancestor from the families carrying that rearrangement. After performing the haplotype reconstruction in the 11 informative families and assuming a generation time of 25 years, we estimate the age of the c.156_157insAlu BRCA2 mutation to be 558 ± 215 years, that is, most likely well after Portugal became politically independent (in 1143). Our estimate is consistent with the widespread distribution of the mutation in Portugal [5, 6], the country demographic history (the North has been and still is consistently the source of migrants to the South), its occasional finding in countries with strong Portuguese immigration, and with its absence in the other populations studied (e.g., absence of the mutation in Spain, namely in Galicia). Nevertheless, statistical methods for estimating mutation ages are relatively crude [16], are dependent on sample representativeness, and estimate only the age of the common ancestor to the informative families that have been identified. The older age estimate advanced by Machado et al. [5] was based upon a different sample of Portuguese patients (mostly from Center and South) and using a different age estimate method. However these authors recognize that the age of the mutation may be «overestimated, either because of the fact that mutation rates of the microsatellite markers were not taken into account or because recombination events in two families were considered». On the other hand, although the mutation has so far only been detected in Portugal and in a few families with Portuguese ancestry living in Belgium, France or the USA, we can not conclusively exclude its presence in other countries that have strong historical links with Portugal, such as those having Portuguese as official language (Brazil, Angola, Mozambique, Cape Verde, Guinea-Bissau, São Tomé and Príncipe, East Timor, and Macau) or other countries with a large community of Portuguese immigrants. In fact, one of our probands with the c.156_157insAlu BRCA2 mutation illustrates this possibility: although she is now living in Portugal, her ancestors originating from North Portugal had moved several generations ago to Brazil and later to Angola, where reportedly various affected relatives lived.
In conclusion, we showed that the c.156_157insAlu BRCA2 rearrangement is a Portuguese founder mutation originated about 558 ± 215 years ago, accounting for the majority of the BRCA2 mutations and for about one-third of all deleterious germline mutations in Portuguese HBOC families. We therefore recommend that all suspected HBOC families from Portugal or with Portuguese ancestry are specifically tested for this rearrangement, ideally prior to screening the entire coding regions of BRCA1 and BRCA2. We further showed that the cumulative incidence of breast cancer in c.156_157insAlu BRCA2 mutation carriers does not differ from that of other BRCA2 and BRCA1 pathogenic mutations and that this BRCA2 rearrangement causes hereditary breast/ovarian cancer because the mutated allele is only able to give rise to BRCA2-Δex3 transcripts and not to BRCA2-wt transcripts.
References
Ferla R, Calo V, Cascio S, Rinaldi G, Badalamenti G, Carreca I, Surmacz E, Colucci G, Bazan V, Russo A (2007) Founder mutations in BRCA1 and BRCA2 genes. Ann Oncol 18:93–98
Fackenthal JD, Olopade OI (2007) Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat Rev Cancer 7(12):937–948
Filippini S, Blanco A, Fernandez-Marmiesse A, Alvarez-Iglesias V, Ruiz-Ponte C, Carracedo A, Vega A (2007) Multiplex SNaPshot for detection of BRCA1/2 common mutations in Spanish and Spanish related breast/ovarian cancer families BMC. Med Genet 8:40
Teugels E, De Brakeleer S, Goelen G, Lissens W, Sermijn E, De Greve J (2005) De novo Alu element insertions targeted to a sequence common to the BRCA1 and BRCA2 genes. Hum Mutat 26(3):284
Machado PM, Brandao RD, Cavaco BM, Eugenio J, Bento S, Nave M, Rodrigues P, Fernandes A, Vaz F (2007) Screening for a BRCA2 rearrangement in high-risk breast/ovarian cancer families: evidence for a founder effect and analysis of the associated phenotypes. J Clin Oncol 25(15):2027–2034
Peixoto A, Santos C, Rocha P, Pinheiro M, Principe S, Pereira D, Rodrigues H, Castro F, Abreu J, Gusmao L, Amorim A, Teixeira MR (2009) The c.156_157insAlu BRCA2 rearrangement accounts for more than one-fourth of deleterious BRCA mutations in northern/central. Portugal Breast Cancer Res Treat 114(1):31–38
Diez O, Gutierrez-Enriquez S, Ramon y Cajal T, Alonso C, Balmana J, Llort G (2007) Caution should be used when interpreting alterations affecting the exon 3 of the BRCA2 gene in breast/ovarian cancer families. J Clin Oncol 25(31):5035–5036
Peixoto A, Santos C, Rocha P, Pinto P, Bizarro S, Teixeira MR (2009) Molecular diagnosis of the Portuguese founder mutation BRCA2 c.156_157insAlu. Breast Cancer Res Treat 117(1):215–217
Peixoto A, Salgueiro N, Santos C, Varzim G, Rocha P, Soares MJ, Pereira D, Rodrigues H, Bento MJ, Fraguas A, Moura G, Regateiro F, Castedo S, Teixeira MR (2006) BRCA1 and BRCA2 germline mutational spectrum and evidence for genetic anticipation in Portuguese breast/ovarian cancer families. Fam Cancer 5(4):379–387
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3(6):1101–1108
Martins S, Calafell F, Gaspar C, Wong VC, Silveira I, Nicholson GA, Brunt ER, Tranebjaerg L, Stevanin G, Hsieh M, Soong BW, Loureiro L, Durr A, Tsuji S, Watanabe M, Jardim LB, Giunti P, Riess O, Ranum LP, Brice A, Rouleau GA, Coutinho P, Amorim A, Sequeiros J (2007) Asian origin for the worldwide-spread mutational event in Machado-Joseph disease. Arch Neurol 64(10):1502–1508
Gyapay G, Morissette J, Vignal A, Dib C, Fizames C, Millasseau P, Marc S, Bernardi G, Lathrop M, Weissenbach J (1994) The 1993–94 Genethon human genetic linkage map. Nat Genet 7. doi:10.1038/ng0694supp-246
Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, Bishop DT, Weber B, Lenoir G, Chang-Claude J, Sobol H, Teare MD, Struewing J, Arason A, Scherneck S, Peto J, Rebbeck TR, Tonin P, Neuhausen S, Barkardottir R, Eyfjord J, Lynch H, Ponder BA, Gayther SA, Zelada-Hedman M et al (1998) Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 62(3):676–689
Santos C, Peixoto A, Rocha P, Vega A, Soares MJ, Cerveira N, Bizarro S, Pinheiro M, Pereira D, Rodrigues H, Castro F, Henrique R, Teixeira MR (2009) Haplotype and quantitative transcript analyses of Portuguese breast/ovarian cancer families with the BRCA1 R71G founder mutation of Galician origin. Fam Cancer 8(3):203–208
McClellan J, King MC (2010) Genetic heterogeneity in human disease. Cell 141(2):210–217
Rannala B, Bertorelle G (2001) Using linked markers to infer the age of a mutation. Hum Mutat 18(2):87–100
Acknowledgments
This study was supported by Ministério da Saúde (Project Nº 15/2007) and Liga Portuguesa Contra o Cancro. IPATIMUP was funded by Fundação para a Ciência e Tecnologia, through POCI (Programa Operacional Ciência e Inovação 2010). MT and WDF are supported by the Susan G. Komen Foundation for the Cure, the Jewish General Hospital Weekend to End Breast Cancer and the Fonds de la Recherche en Santé du Québec. EAV and MD are supported by grants PI061102 (Ministerio de Ciencia e Innovación) and 200820I135 (CSIC). AV and AB thank María J. Magdalena for her excellent technical support at the Fundación Pública Galega de Medicina Xenómica. PAP and IPE were supported by CNPq, CAPES, Fundação de Incentivo à Pesquisa do Hospital de Clínicas de Porto Alegre, and Rede Nacional de Câncer Familial, Brazil. The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary file
DOC 91 kb
Supplementary Fig. 1
Map of Portugal showing the known geographical origin of the families with the c.156_157insAlu BRCA2 germline mutation. The black circles indicate the origin of the 25 families detected in Portugal (present report and those reported in Peixoto et al. [6]), the open circle the origin of the family previously identified by Teugels et al. [4] in Belgium, and the triangles the origin of the families of the four individuals (two from each) subjected to predictive testing living in Rhode Island, USA, and in Villejuif, France, respectively. (TIF 4,506 kb)
Supplementary Fig. 2
SNP marker haplotype spanning ~1.1Mb, encompassing the region between the D13S260 and D13S1695 microsatellite markers, in the 11 informative families. (TIF 7,026 kb)
Rights and permissions
About this article
Cite this article
Peixoto, A., Santos, C., Pinheiro, M. et al. International distribution and age estimation of the Portuguese BRCA2 c.156_157insAlu founder mutation. Breast Cancer Res Treat 127, 671–679 (2011). https://doi.org/10.1007/s10549-010-1036-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-010-1036-3