
Abstract
Published data on the association between MTHFR C677T polymorphism and breast cancer risk are inconclusive. To derive a more precise estimation of the relationship, a meta-analysis was performed. Medline, PubMed, Embase, and Web of Science were searched. Crude ORs with 95% CIs were used to assess the strength of association between the MTHFR C677T polymorphism and breast cancer risk. The pooled ORs were performed with co-dominant model (CT vs. CC, TT vs. CC), dominant model (CT + TT vs. CC), and recessive model (TT vs. CC + CT), respectively. A total of 37 studies including 15,260 cases and 20,411 controls were involved in this meta-analysis. Overall, significantly elevated breast cancer risk was associated with TT variant genotype in homozygote comparison and dominant genetic model when all studies were pooled into the meta-analysis (TT vs. CC: OR = 1.11, 95% CI = 1.01–1.23; dominant model: OR = 1.04, 95% CI = 1.00–1.09). In the subgroup analysis by ethnicity, significantly increased risks were found for TT allele carriers among Asians (TT vs. CC: OR = 1.18, 95% CI = 1.04–1.35; recessive model: OR = 1.15, 95% CI = 1.03–1.29). When stratified by study design, statistically significantly elevated risk was found in hospital-based studies (TT vs. CC: OR = 1.18, 95% CI = 1.02–1.38; recessive model: OR = 1.17, 95% CI = 1.05–1.29). In the subgroup analysis by menopausal status, statistically significantly increased risk was found among postmenopausal women (CT vs. CC: OR = 1.12, 95% CI = 1.02–1.23; dominant model: OR = 1.11, 95% CI = 1.01–1.22). In conclusion, this meta-analysis suggests that the MTHFR T allele is a low-penetrant risk factor for developing breast cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In 2009, breast cancer was expected to account for 27% (192,370) of all new cancer cases among women in the United States [1]. Breast cancer is still a major challenge for women’s health. While the exact etiology of the disease is poorly understood, there are some recognized risk factors that may contribute to the development of breast cancer including age, ethnicity, reproductive events, exogenous hormones, lifestyle, bone density, as well as genetic factors [2]. It has been suggested that low-penetrance susceptibility genes combining with environmental factors may be important in the development of cancer [3]. In recent years, several common low-penetrant genes have been identified as potential breast cancer susceptibility genes, one of which is 5,10-methylenetetrahydrofolate reductase (MTHFR) gene. It encodes a critical enzyme for intracellular folate homeostasis and metabolism, which catalyzes the conversion of 5,10-methylenetetrahydrofolate (5,10-methylene-THF) to 5-methyltetrahydrofolate (5-methylene-THF). The latter is the predominant form of folate in plasma and provides the methyl group for de novo methionine synthesis through homocysteine remethylation [4]. A common polymorphism in the MTHFR gene has been identified in which a C to T substitution at nucleotide position 677 (C677T) results in an alanine to valine substitution and the production of a thermolabile variant of the MTHFR enzyme that has approximately 30% of the activity of the wild-type enzyme [5, 6]. This polymorphism to breast cancer risk has been a research focus in scientific community and has drawn increasing attention. Forty original studies have reported the role of MTHFR C677T polymorphism in breast cancer risk [7–46], but the results are inconclusive, partially because of the possible small effect of the polymorphism on breast cancer risk and the relatively small sample size in each of published studies. Therefore, we performed this meta-analysis to derive a more precise estimation of these associations.
Methods
Publication search
Medline, PubMed, Embase, and Web of Science were searched (last search was done on Jan 22, 2010, using the search terms: “methylenetetrahydrofolate reductase,” “MTHFR,” “polymorphism,” and “breast”). All searched studies were retrieved, and their bibliographies were checked for other relevant publications. Review articles and bibliographies of other relevant studies identified were hand-searched to find additional eligible studies. Only published studies with full text articles were included. When more than one of the same patient population was included in several publications, only the most recent or complete study was used in this meta-analysis.
Inclusion criteria
The inclusion criteria were: (a) evaluation of the MTHFR C677T polymorphism and breast cancer risk, (b) case–control studies, and (c) sufficient published data for estimating an odds ratio (OR) with 95% confidence interval (CI).
Data extraction
Information was carefully extracted from all eligible publications independently by two of the authors according to the inclusion criteria listed above. Disagreement was resolved by discussion between the two authors. If these two authors could not reach a consensus, another author was consulted to resolve the dispute and a final decision was made by the majority of the votes. The following data were collected from each study: first author’s name, publication date, ethnicity, study design, menopausal status, total number of cases and controls, and numbers of cases and controls with the MTHFR C677T genotypes, respectively. Different ethnicities were categorized as Caucasian, Asian, mixed or not-stated. Study design was stratified into population-based studies and hospital-based studies. Menopausal status was divided into premenopausal and postmenopausal. We did not define any minimum number of patients to be included in our meta-analysis.
Statistical methods
Crude ORs with 95% CIs were used to assess the strength of association between the MTHFR C677T polymorphism and breast cancer risk. The pooled ORs were performed with co-dominant model (CT vs. CC, TT vs. CC), dominant model (CT + TT vs. CC), and recessive model (TT vs. CC + CT), respectively. Heterogeneity assumption was checked with the Chi-square-based Q-test [47]. A P value greater than 0.10 for the Q-test indicates a lack of heterogeneity among studies, so the pooled OR estimate of the each study was calculated with the fixed-effects model (the Mantel–Haenszel method) [48]. Otherwise, the random-effects model (the DerSimonian and Laird method) was used [49]. Subgroup analyses were performed by ethnicity, study design, and menopausal status. Sensitivity analysis was performed to assess the stability of the results. A single study involved in the meta-analysis was deleted each time to define the influence of the individual data-set to the pooled ORs [50]. An estimate of potential publication bias was carried out by the funnel plot, in which the standard error of log (OR) of each study was plotted against its log (OR). An asymmetric plot suggests a possible publication bias. Funnel plot asymmetry was assessed by the method of Egger’s linear regression test, a linear regression approach to measure funnel plot asymmetry on the natural logarithm scale of the OR. The significance of the intercept was determined with the t-test suggested by Egger (P < 0.05 was considered representative of statistically significant publication bias) [51]. All the statistical tests were performed with STATA version 10.0 (Stata Corporation, College Station, TX).
Results
Study characteristics
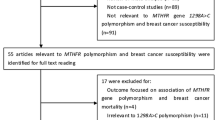
A total of 40 publications met the inclusion criteria [7–46]. Studies of Ericson et al. [16], Tao et al. [44], and Xu et al. [45] were excluded because the subjects had been included by Ericson et al. [15], Platek et al. [35], and Chen et al. [10], respectively. Hence, a total of 37 studies including 15,260 cases and 20,411 controls were used in the meta-analysis. Table 1 lists the studies identified and their main characteristics. Of the 37 studies, sample sizes ranged from 93 to 4,256. There were 13 studies of Caucasians, 12 studies of Asians, 8 studies of mixed populations, and 4 studies of unstated populations. Almost all of the cases were pathologically confirmed. Controls were mainly healthy populations and matched for age. Among these studies, 15 were population-based and 22 were hospital-based.
Main results
Table 2 lists the main results of this meta-analysis. Overall, significantly elevated breast cancer risk was associated with MTHFR T allele when all studies were pooled into the meta-analysis (TT vs. CC: OR = 1.11, 95% CI = 1.01–1.23; dominant model: OR = 1.04, 95% CI = 1.00–1.09). In the subgroup analysis by ethnicity, significantly increased risk was only found for Asians (TT vs. CC: OR = 1.18, 95% CI = 1.04–1.35; recessive model: OR = 1.15, 95% CI = 1.03–1.29). When stratified by study design, statistically significantly elevated risk was found in hospital-based studies (TT vs. CC: OR = 1.18, 95% CI = 1.02–1.38; recessive model: OR = 1.17, 95% CI = 1.05–1.29). In the subgroup analysis by menopausal status, statistically significantly increased risk was found among postmenopausal women (CT vs. CC: OR = 1.12, 95% CI = 1.02–1.23; dominant model: OR = 1.11, 95% CI = 1.01–1.22).
Sensitivity analysis
A single study involved in the meta-analysis was deleted each time to define the influence of the individual data-set to the pooled ORs, and the corresponding pooled ORs were not materially altered (data not shown), indicating that our results were statistically robust.
Publication bias
Begg’s funnel plot and Egger’s test were performed to assess the publication bias of literatures. The shape of the funnel plot did not reveal any evidence of obvious asymmetry (figures not shown). Then, the Egger’s test was used to provide statistical evidence of funnel plot symmetry. The results still did not suggest any evidence of publication bias (P = 0.76 for CT vs. CC; P = 0.14 for TT vs. CC; P = 0.32 with dominant model; and P = 0.16 with recessive model).
Discussion
The present meta-analysis, including 15,260 cases and 20,411 controls, explored the association between the MTHFR C677T polymorphism and breast cancer risk. The results from our meta-analysis indicate that the MTHFR T allele is a low-penetrant risk factor for developing breast cancer.
This finding may be biologically plausible. Individuals with the MTHFR 677TT genotype have been shown to have only 30% of in vitro MTHFR enzyme activity compared with the wild type, whereas those with the heterozygous CT genotype have 60% of wild-type MTHFR enzyme activity [5]. Reduction of the MTHFR enzyme activity increases the pool of 5,10-methylene-THF at the expense of the pool of 5-methyl-THF, which impairs the DNA methylation. DNA methylation plays a critical role in regulation of gene expression and maintenance of genomic stability [52, 53] and aberrations in normal methylation patterns have been associated with the development of cancer [53, 54]. More importantly, the homozygous variant genotype MTHFR 677TT has been associated with risk for many different types of cancer, including colorectal [55], gastric [56], endometrial [57], lung cancer [58], and acute leukemia [59].
In the subgroup analysis based on ethnicities, significant associations were found in Asians but not for Caucasians under TT vs. CC and with recessive genetic models, suggesting a possible role of ethnic differences in genetic backgrounds and the environment they live in [60].
Our results showed that significantly increased breast cancer risk in MTHFR T genotype carriers were found in the hospital-based studies but not in population-based studies. The hospital-based studies usually have some biases because such controls may just represent a sample of ill-defined reference population, and may not be representative of the general population very well, particularly when the genotypes under investigation were associated with the disease conditions that the hospital-based controls may have. If considering this kind of selection bias, our results should be interpreted with caution.
When stratified by menopausal status, a more pronounced increased breast cancer risk was observed in postmenopausal women under CT vs. CC and with dominant genetic models. Though results from available case–control studies [7–46] investigating MTHFR C677T and breast cancer risk have been inconsistent, our results are consistent with three of them reporting significant positive associations for postmenopausal women. Ericson et al. observed a significant 34% increase in breast cancer risk among postmenopausal women in Sweden with CT and TT genotypes compared to wild type in a nested case–control study of the Malmo Diet and Cancer cohort [15]. Suzuki reported a significant 83% increased breast cancer risk among postmenopausal Japanese women with the TT genotype compared to wild type [43]. Maruti et al. observed a 62% increased risk of breast cancer among postmenopausal women with the TT genotype [34]. But other investigations have not reported this kind of significant associations. Differences in results may be due to variation between populations with regards to prevalence of polymorphisms in genes related to one-carbon metabolism, intakes of nutrients, and/or other risk factors for breast cancer. Data from future in-depth research on these gene–gene or gene–environment interactions may further elucidate this issue.
Some limitations of this meta-analysis should be acknowledged. Firstly, the controls were not uniformly defined. Although most of the controls were selected mainly from healthy populations, some had benign disease. Therefore, non-differential misclassification bias was possible because these studies may have included the control groups who have different risks of developing breast cancer. Secondly, some studies with small sample size may not have enough statistical power to explore the real association. Thirdly, our results were based on unadjusted estimates, while a more precise analysis should be conducted if all individual data were available, which would allow for the adjustment by other co-variants including age, ethnicity, smoking status, drinking status, obesity, environmental factors, and other lifestyle. In spite of these limitations, our meta-analysis had several strengths. First, substantial number of cases and controls were pooled from different studies, which significantly increased the statistical power of the analysis. Second, no publication biases were detected, indicating that the whole pooled results may be unbiased.
In conclusion, this meta-analysis suggests that the MTHFR T allele is a low-penetrant risk factor for developing breast cancer. However, it is necessary to conduct large sample studies using standardized unbiased genotyping methods, homogeneous breast cancer patients and well matched controls. Moreover, gene–gene and gene–environment interactions should also be considered in the analysis. Such studies taking these factors into account may eventually lead to our better, comprehensive understanding of the association between the MTHFR C677T polymorphism and breast cancer risk.
References
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ (2009) Cancer statistics, 2009. CA Cancer J Clin 59:225–249
Dumitrescu RG, Cotarla I (2005) Understanding breast cancer risk—where do we stand in 2005? J Cell Mol Med 9:208–221
Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K (2000) Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 343:78–85
Bailey LB, Gregory JR (1999) Polymorphisms of methylenetetrahydrofolate reductase and other enzymes: metabolic significance, risks and impact on folate requirement. J Nutr 129:919–922
Frosst P, Blom HJ, Milos R, Goyette P, Sheppard CA, Matthews RG, Boers GJ, den Heijer M, Kluijtmans LA, van den Heuvel LP, Et A (1995) A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet 10:111–113
Jacques PF, Bostom AG, Williams RR, Ellison RC, Eckfeldt JH, Rosenberg IH, Selhub J, Rozen R (1996) Relation between folate status, a common mutation in methylenetetrahydrofolate reductase, and plasma homocysteine concentrations. Circulation 93:7–9
Kalyankumar Ch, Jamil K (2006) Methylene tetrahydofolate reductase (MTHFR) C677T and A1298C polymorphisms and breast cancer in South Indian population. Int J Cancer Res 2:143–151
Cam R, Eroglu A, Egin Y, Akar N (2009) Dihydrofolate reductase (DHRF) 19-bp intron-1 deletion and methylenetetrahydrofolate reductase (MTHFR) C677T polymorphisms in breast cancer. Breast Cancer Res Treat 115:431–432
Campbell IG, Baxter SW, Eccles DM, Choong DY (2002) Methylenetetrahydrofolate reductase polymorphism and susceptibility to breast cancer. Breast Cancer Res 4:R14
Chen J, Gammon MD, Chan W, Palomeque C, Wetmur JG, Kabat GC, Teitelbaum SL, Britton JA, Terry MB, Neugut AI, Santella RM (2005) One-carbon metabolism, MTHFR polymorphisms, and risk of breast cancer. Cancer Res 65:1606–1614
Cheng CW, Yu JC, Huang CS, Shieh JC, Fu YP, Wang HW, Wu PE, Shen CY (2008) Polymorphism of cytosolic serine hydroxymethyltransferase, estrogen and breast cancer risk among Chinese women in Taiwan. Breast Cancer Res Treat 111:145–155
Chou YC, Wu MH, Yu JC, Lee MS, Yang T, Shih HL, Wu TY, Sun CA (2006) Genetic polymorphisms of the methylenetetrahydrofolate reductase gene, plasma folate levels and breast cancer susceptibility: a case-control study in Taiwan. Carcinogenesis 27:2295–2300
Deligezer U, Akisik EE, Dalay N (2005) Homozygosity at the C677T of the MTHFR gene is associated with increased breast cancer risk in the Turkish population. In Vivo 19:889–893
Ergul E, Sazci A, Utkan Z, Canturk NZ (2003) Polymorphisms in the MTHFR gene are associated with breast cancer. Tumour Biol 24:286–290
Ericson U, Sonestedt E, Ivarsson MI, Gullberg B, Carlson J, Olsson H, Wirfalt E (2009) Folate intake, methylenetetrahydrofolate reductase polymorphisms, and breast cancer risk in women from the Malmo Diet and Cancer cohort. Cancer Epidemiol Biomarkers Prev 18:1101–1110
Ericson UC, Ivarsson MI, Sonestedt E, Gullberg B, Carlson J, Olsson H, Wirfalt E (2009) Increased breast cancer risk at high plasma folate concentrations among women with the MTHFR 677T allele. Am J Clin Nutr 90:1380–1389
Forsti A, Angelini S, Festa F, Sanyal S, Zhang Z, Grzybowska E, Pamula J, Pekala W, Zientek H, Hemminki K, Kumar R (2004) Single nucleotide polymorphisms in breast cancer. Oncol Rep 11:917–922
Gao CM, Tang JH, Cao HX, Ding JH, Wu JZ, Wang J, Liu YT, Li SP, Su P, Matsuo K, Takezaki T, Tajima K (2009) MTHFR polymorphisms, dietary folate intake and breast cancer risk in Chinese women. J Hum Genet 54:414–418
Grieu F, Powell B, Beilby J, Iacopetta B (2004) Methylenetetrahydrofolate reductase and thymidylate synthase polymorphisms are not associated with breast cancer risk or phenotype. Anticancer Res 24:3215–3219
Hekim N, Ergen A, Yaylim I, Yilmaz H, Zeybek U, Ozturk O, Isbir T (2007) No association between methylenetetrahydrofolate reductase C677T polymorphism and breast cancer. Cell Biochem Funct 25:115–117
Henriquez-Hernandez LA, Murias-Rosales A, Hernandez GA, Cabrera DLA, Diaz-Chico BN, Mori DSM, Fernandez PL (2009) Gene polymorphisms in TYMS, MTHFR, p53 and MDR1 as risk factors for breast cancer: a case-control study. Oncol Rep 22:1425–1433
Inoue M, Robien K, Wang R, Van Den Berg DJ, Koh WP, Yu MC (2008) Green tea intake, MTHFR/TYMS genotype and breast cancer risk: the Singapore Chinese Health Study. Carcinogenesis 29:1967–1972
Justenhoven C, Hamann U, Pierl CB, Rabstein S, Pesch B, Harth V, Baisch C, Vollmert C, Illig T, Bruning T, Ko Y, Brauch H (2005) One-carbon metabolism and breast cancer risk: no association of MTHFR, MTR, and TYMS polymorphisms in the GENICA study from Germany. Cancer Epidemiol Biomarkers Prev 14:3015–3018
Kalemi TG, Lambropoulos AF, Gueorguiev M, Chrisafi S, Papazisis KT, Kotsis A (2005) The association of p53 mutations and p53 codon 72, Her 2 codon 655 and MTHFR C677T polymorphisms with breast cancer in Northern Greece. Cancer Lett 222:57–65
Kotsopoulos J, Zhang WW, Zhang S, McCready D, Trudeau M, Zhang P, Sun P, Narod SA (2008) Polymorphisms in folate metabolizing enzymes and transport proteins and the risk of breast cancer. Breast Cancer Res Treat 112:585–593
Langsenlehner U, Krippl P, Renner W, Yazdani-Biuki B, Wolf G, Wascher TC, Paulweber B, Weitzer W, Samonigg H (2003) The common 677C > T gene polymorphism of methylenetetrahydrofolate reductase gene is not associated with breast cancer risk. Breast Cancer Res Treat 81:169–172
Le Marchand L, Haiman CA, Wilkens LR, Kolonel LN, Henderson BE (2004) MTHFR polymorphisms, diet, HRT, and breast cancer risk: the multiethnic cohort study. Cancer Epidemiol Biomarkers Prev 13:2071–2077
Lee SA, Kang D, Nishio H, Lee MJ, Kim DH, Han W, Yoo KY, Ahn SH, Choe KJ, Hirvonen A, Noh DY (2004) Methylenetetrahydrofolate reductase polymorphism, diet, and breast cancer in Korean women. Exp Mol Med 36:116–121
Lin WY, Chou YC, Wu MH, Huang HB, Jeng YL, Wu CC, Yu CP, Yu JC, You SL, Chu TY, Chen CJ, Sun CA (2004) The MTHFR C677T polymorphism, estrogen exposure and breast cancer risk: a nested case-control study in Taiwan. Anticancer Res 24:3863–3868
Lissowska J, Gaudet MM, Brinton LA, Chanock SJ, Peplonska B, Welch R, Zatonski W, Szeszenia-Dabrowska N, Park S, Sherman M, Garcia-Closas M (2007) Genetic polymorphisms in the one-carbon metabolism pathway and breast cancer risk: a population-based case-control study and meta-analyses. Int J Cancer 120:2696–2703
Ma E, Iwasaki M, Junko I, Hamada GS, Nishimoto IN, Carvalho SM, Motola JJ, Laginha FM, Tsugane S (2009) Dietary intake of folate, vitamin B6, and vitamin B12, genetic polymorphism of related enzymes, and risk of breast cancer: a case-control study in Brazilian women. BMC Cancer 9:122
Ma E, Iwasaki M, Kobayashi M, Kasuga Y, Yokoyama S, Onuma H, Nishimura H, Kusama R, Tsugane S (2009) Dietary intake of folate, vitamin B2, vitamin B6, vitamin B12, genetic polymorphism of related enzymes, and risk of breast cancer: a case-control study in Japan. Nutr Cancer 61:447–456
Macis D, Maisonneuve P, Johansson H, Bonanni B, Botteri E, Iodice S, Santillo B, Penco S, Gucciardo G, D’Aiuto G, Rosselli DTM, Amadori M, Costa A, Decensi A (2007) Methylenetetrahydrofolate reductase (MTHFR) and breast cancer risk: a nested-case-control study and a pooled meta-analysis. Breast Cancer Res Treat 106:263–271
Maruti SS, Ulrich CM, Jupe ER, White E (2009) MTHFR C677T and postmenopausal breast cancer risk by intakes of one-carbon metabolism nutrients: a nested case-control study. Breast Cancer Res 11:R91
Platek ME, Shields PG, Marian C, McCann SE, Bonner MR, Nie J, Ambrosone CB, Millen AE, Ochs-Balcom HM, Quick SK, Trevisan M, Russell M, Nochajski TH, Edge SB, Freudenheim JL (2009) Alcohol consumption and genetic variation in methylenetetrahydrofolate reductase and 5-methyltetrahydrofolate-homocysteine methyltransferase in relation to breast cancer risk. Cancer Epidemiol Biomarkers Prev 18:2453–2459
Qi J, Miao XP, Tan W, Yu CY, Liang G, Lu WF, Lin DX (2004) Association between genetic polymorphisms in methylenetetrahydrofolate reductase and risk of breast cancer. Zhonghua Zhong Liu Za Zhi 26:287–289
Reljic A, Simundic AM, Topic E, Nikolac N, Justinic D, Stefanovic M (2007) The methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism and cancer risk: the Croatian case-control study. Clin Biochem 40:981–985
Semenza JC, Delfino RJ, Ziogas A, Anton-Culver H (2003) Breast cancer risk and methylenetetrahydrofolate reductase polymorphism. Breast Cancer Res Treat 77:217–223
Sharp L, Little J, Schofield AC, Pavlidou E, Cotton SC, Miedzybrodzka Z, Baird JO, Haites NE, Heys SD, Grubb DA (2002) Folate and breast cancer: the role of polymorphisms in methylenetetrahydrofolate reductase (MTHFR). Cancer Lett 181:65–71
Shrubsole MJ, Gao YT, Cai Q, Shu XO, Dai Q, Hebert JR, Jin F, Zheng W (2004) MTHFR polymorphisms, dietary folate intake, and breast cancer risk: results from the Shanghai Breast Cancer Study. Cancer Epidemiol Biomarkers Prev 13:190–196
Sohn KJ, Croxford R, Yates Z, Lucock M, Kim YI (2004) Effect of the methylenetetrahydrofolate reductase C677T polymorphism on chemosensitivity of colon and breast cancer cells to 5-fluorouracil and methotrexate. J Natl Cancer Inst 96:134–144
Stevens VL, McCullough ML, Pavluck AL, Talbot JT, Feigelson HS, Thun MJ, Calle EE (2007) Association of polymorphisms in one-carbon metabolism genes and postmenopausal breast cancer incidence. Cancer Epidemiol Biomarkers Prev 16:1140–1147
Suzuki T, Matsuo K, Hirose K, Hiraki A, Kawase T, Watanabe M, Yamashita T, Iwata H, Tajima K (2008) One-carbon metabolism-related gene polymorphisms and risk of breast cancer. Carcinogenesis 29:356–362
Tao MH, Shields PG, Nie J, Marian C, Ambrosone CB, McCann SE, Platek M, Krishnan SS, Xie B, Edge SB, Winston J, Vito D, Trevisan M, Freudenheim JL (2009) DNA promoter methylation in breast tumors: no association with genetic polymorphisms in MTHFR and MTR. Cancer Epidemiol Biomarkers Prev 18:998–1002
Xu X, Gammon MD, Zhang H, Wetmur JG, Rao M, Teitelbaum SL, Britton JA, Neugut AI, Santella RM, Chen J (2007) Polymorphisms of one-carbon-metabolizing genes and risk of breast cancer in a population-based study. Carcinogenesis 28:1504–1509
Yu CP, Wu MH, Chou YC, Yang T, You SL, Chen CJ, Sun CA (2007) Breast cancer risk associated with multigenotypic polymorphisms in folate-metabolizing genes: a nested case-control study in Taiwan. Anticancer Res 27:1727–1732
Cochran WG (1954) The combination of estimates from different experiments. Biometrics 10:101–129
Mantel N, Haenszel W (1959) Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 22:719–748
DerSimonian R, Laird N (1986) Meta-analysis in clinical trials. Control Clin Trials 7:177–188
Tobias A (1999) Assessing the influence of a single study in the meta-analysis estimate. Stata Tech Bull 8:15–17
Egger M, Davey Smith G, Schneider M, Minder C (1997) Bias in meta-analysis detected by a simple, graphical test. BMJ 315:629–634
Kundu TK, Rao MR (1999) CpG islands in chromatin organization and gene expression. J Biochem 125:217–222
Lengauer C, Kinzler KW, Vogelstein B (1997) DNA methylation and genetic instability in colorectal cancer cells. Proc Natl Acad Sci USA 94:2545–2550
Cheng P, Schmutte C, Cofer KF, Felix JC, Yu MC, Dubeau L (1997) Alterations in DNA methylation are early, but not initial, events in ovarian tumorigenesis. Br J Cancer 75:396–402
Ma J, Stampfer MJ, Giovannucci E, Artigas C, Hunter DJ, Fuchs C, Willett WC, Selhub J, Hennekens CH, Rozen R (1997) Methylenetetrahydrofolate reductase polymorphism, dietary interactions, and risk of colorectal cancer. Cancer Res 57(6):1098–1102
Graziano F, Kawakami K, Ruzzo A, Watanabe G, Santini D, Pizzagalli F, Bisonni R, Mari D, Floriani I, Catalano V, Silva R, Tonini G, Torri V, Giustini L, Magnani M (2006) Methylenetetrahydrofolate reductase 677C/T gene polymorphism, gastric cancer susceptibility and genomic DNA hypomethylation in an at-risk Italian population. Int J Cancer 118(3):628–632
Esteller M, Garcia A, Martinez-Palones JM, Xercavins J, Reventos J (1997) Germ line polymorphisms in cytochrome-P450 1A1 (C4887 CYP1A1) and methylenetetrahydrofolate reductase (MTHFR) genes and endometrial cancer susceptibility. Carcinogenesis 18(12):2307–2311
Siemianowicz K, Gminski J, Garczorz W, Slabiak N, Goss M, Machalski M, Magiera-Molendowska H (2003) Methylenetetrahydrofolate reductase gene C677T and A1298C polymorphisms in patients with small cell and non-small cell lung cancer. Oncol Rep 10(5):1341–1344
Skibola CF, Smith MT, Kane E, Roman E, Rollinson S, Cartwright RA, Morgan G (1999) Polymorphisms in the methylenetetrahydrofolate reductase gene are associated with susceptibility to acute leukemia in adults. Proc Natl Acad Sci USA 96(22):12810–12815
Hirschhorn JN, Lohmueller K, Byrne E (2002) A comprehensive review of genetic association studies. Genet Med 4:45–61
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, J., Qiu, LX., Wang, ZH. et al. MTHFR C677T polymorphism associated with breast cancer susceptibility: a meta-analysis involving 15,260 cases and 20,411 controls. Breast Cancer Res Treat 123, 549–555 (2010). https://doi.org/10.1007/s10549-010-0783-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-010-0783-5