
Abstract
The purpose of this study was to determine the safety and maximum tolerated dose (MTD) of BZL101 (FDA IND# 59,521), an orally delivered aqueous extract from the herb Scutellaria barbata, in women with metastatic breast cancer (MBC). The trial was an open-label, phase 1B, multicenter, dose escalation study. Eligible patients had histologically confirmed breast cancer and measurable stage IV disease. The standard phase 1 “3 + 3” study design was used to determine the MTD. Primary endpoints were toxicity and MTD of BZL101. Secondary outcomes included efficacy based on RECIST criteria. A total of 27 women with a median of 2 prior chemotherapy treatments for metastatic disease were treated in four different dose cohorts. Grade 3 and 4 adverse events (AEs) were uncommon. Dose-limiting toxicities included the following: grade 4 AST elevation, grade 3 diarrhea, grade 3 fatigue, and grade 3 rib pain. Fourteen patients were evaluable according to Response Evaluation Criteria in Solid Tumors. Investigator assessment classified three patients with stable disease for >120 days (21%). One patient was on BZL101 for 449 days and remains stable for 700 + days. Independent radiology review identified three patients with objective tumor regression (>0% and <30%). The MTD was not reached, thus per protocol, the MTD was defined as the maximum administered dose of BZL101 40 g/day. In conclusion, oral administration of BZL101 was safe, well tolerated, and showed promising clinical evidence of anticancer activity in this heavily pretreated population of women with MBC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the United States, it is estimated that over 192,000 women will be diagnosed with breast cancer in 2009 and over 40,000 women will die from the disease [1]. While advances in early detection and adjuvant therapy have had a favorable impact on survival, locally advanced or metastatic breast cancer (MBC) remains incurable. Commonly used hormonal and chemotherapeutic agents can lead to transient regression of tumors and palliate symptoms related to cancer; however, these treatments are often accompanied by significant toxicities and eventually become ineffective in controlling advanced stage breast cancer and its symptoms [2]. Even newer targeted biological agents lead to only modest improvements in survival [3]. Moreover, patients ultimately develop resistance to available treatments, cumulative toxicities, or intolerable side effects. Given that women with MBC already suffer from compromised immune systems as a result of prior cytotoxic treatments, effective novel treatment options with less toxicity and higher tolerability are urgently needed.
BZL101 is an aqueous extract of the aerial part of Scutellaria barbata (Chinese pin yin transliteration: Ban Zhi Lian). The plant, which is grown in China mainly in areas southeast of the Yellow River, is harvested in late summer and early autumn. The herb is identified through botanical, morphological, and chemical characteristics to ensure identity, purity, potency, and consistency of the product according to the Food and Drug Administration’s (FDA) Botanical Drug Guidance. Several flavonoids with cytotoxic activities have been identified and purified from BZL101. Despite identification of several active chemical compounds from BZL101, none demonstrate more potent cytotoxic activity than the whole plant extract. For this reason, the whole herb extract is being studied instead of one of the isolated chemical compounds.
BZL101 resulted in more than 50% growth inhibition in four of five breast cancer cell lines, representing both estrogen receptor positive and negative phenotypes evaluated in vitro [4]. In contrast, at the same dose, BZL101 did not cause more than 50% growth inhibition of normal human mammary epithelial cells (HuMEC) or normal human fibroblasts [5]. The cytotoxic selectivity of BZL101 for cancer cells as opposed to normal cells is based on the metabolic preferences of tumor cells, namely, intrinsically higher basal levels of reactive oxygen species (ROS) and dependence on glycolysis for energy production (Warburg effect) that make cancer cells significantly more vulnerable to BZL101-induced death [6]. Extensive studies to elucidate the mechanism of action of BZL101 have shown [4–6]: (a) BZL101 induces more ROS, and, correspondingly, greater DNA damage in tumor cells than in normal cells, (b) severe DNA damage in tumor cells leads to the hyperactivation of poly (ADP-ribose) polymerase (PARP) and depletion of its substrate NAD and ATP (PARP substrates), and (c) the depletion of NAD results in the inhibition of glycolysis and further decline in ATP because glycolysis is frequently the only energy-generating mechanism in tumor cells [7]. The collapse of redox and energy status is followed by programmed necrosis of tumor cells while normal cells repair DNA damage and continue to produce energy through mitochondrial respiration.
In a phase 1A trial we previously showed that BZL101 for MBC had very limited toxicity and encouraging clinical activity among women with heavily pretreated MBC [8]. In the phase 1A trial, BZL101 was administered as a liquid extract. Although compliance was good, many women chose to discontinue BZL101 because of its bitter taste. In the Phase 1B trial, BZL101 was administered as a freeze-dried powder with excipients to mask the bitterness of the extract. This paper describes the results of the phase 1B clinical trial of reformulated BZL101 for MBC.
Methods
Eligibility
Patients 18 years or older with histologically confirmed breast cancer and measurable stage IV disease defined by Response Evaluation Criteria in Solid Tumors (RECIST) criteria were eligible for the study. The protocol initially did not limit the number of prior chemotherapy regimens for MBC, however, this was amended after enrollment to the first dose cohort to a limit of no more than three prior chemotherapy regimens for MBC. Patients could have any number of prior adjuvant therapies. Additional inclusion criteria were Eastern Cooperative Oncology Group (ECOG) performance status ≤2, life expectancy >12 weeks, resolution of any significant side effects related to prior anticancer treatment, and adequate organ and bone marrow function measured within 14 days of study treatment (hemoglobin ≥10.0 g/dl; absolute neutrophil count ≥1,500/mm3; platelet count ≥100,000/mm3; total bilirubin ≤1.5 mg/dl; ALT and AST ≤2.5X ULN (liver metastases AST/ALT ≤ 5X ULN); alkaline phosphatase ≤3X ULN (liver or bone metastases alkaline phosphatase ≤5X ULN); serum creatinine <1.5 mg/dl). Exclusion criteria included warfarin use, supplements containing Scutellaria barbata, significant comorbidity, pregnancy or breastfeeding, liver involvement of >50% of liver parenchyma, lymphangitic pulmonary involvement, central nervous system involvement, or spinal cord compression not stabilized by therapy for >3 months, clinically significant gastrointestinal abnormalities or toxicities from prior anticancer treatments.
All patients provided written informed consent. Trial approval was obtained by institutional review boards at all clinical sites according to national and local trial center requirements. This study was performed in accordance with International Conference on Harmonization Good Clinical Practice guidelines, and the Declaration of Helsinki (ClinicalTrials.gov No. NCT00454532).
Study design and treatment
This was a multicenter, open-label, dose escalation phase 1B clinical trial. A second phase 1 trial was conducted with a modified formulation to mask the bitterness of the original drug substance and because the phase 1A was not designed as a dose escalation study. BZL101 was administered orally, once or twice daily, on a continuous basis. The trial was sponsored by Bionovo, Inc., Emeryville, CA. The standard phase 1 “3 + 3” Fibonacci design was used to determine the maximum tolerated dose (MTD) [9]. The starting dose of 10 g/day was equivalent by mass to the dose used in the phase 1A study [8]. The dose escalation schedule for BZL101 treatment is displayed in Table 1.
All adverse events, serious adverse events (SAEs) [10], clinically significant abnormal laboratory findings, and abnormal physical exam findings were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 3.0. Treatment was discontinued for any of the following: disease progression, serious protocol violation, withdrawal of consent, or if the patient decided to discontinue treatment.
Dose-limiting toxicity (DLT) was defined as the occurrence of a grade 3, 4 or 5 toxicity deemed possibly, probably, or definitely related to study medication, or a grade 2 gastrointestinal toxicity lasting for >3 weeks deemed possibly, probably, or definitely related to study medication. Gastrointestinal toxicities included any adverse events that fell into the NCI-CTCAE version 3.0 “Gastrointestinal” category. Cohorts of three patients were recruited and treated at specified doses. If all three patients completed 28 days of therapy without a DLT, the dose was escalated in another cohort of patients. If one of the patients experienced a DLT, then another cohort of three was to be recruited at the same dose level. If two or more of the six patients experienced a DLT, the maximum administered dose (MAD) had been defined and dose escalation was to be stopped. No intra-patient dose escalation was permitted. The MAD was defined as the dose at which at least two of six patients experience a DLT. The MTD was defined as the maximum dose at which no more than one of six patients experienced a DLT. If no MTD was determined, the MTD would be defined as the highest dose tested, BZL101 40 g/day.
Assessments
Pretreatment evaluation consisted of a complete medical history, physical examination, ECOG performance status assessment, vital signs, baseline 12 lead ECG, complete blood counts, serum chemistries, and urinalysis. Pretrial screening was performed within 14 days of trial entry. Baseline radiographic assessment could be performed up to 28 days prior to initiation of therapy. After baseline evaluation, patients were assessed every 4 weeks for adverse events using NCI-CTCAE version 3.0. Tumor measurements by physical examination and appropriate scans were performed every 8 weeks using RECIST criteria. Confirmatory scans to verify a partial or complete response were required 4 weeks following initial documentation of an objective response. Independent radiology review was conducted to confirm clinical responses.
Statistical methods
The primary outcomes of the study were the MTD of BZL101 and toxicities associated with treatment. Secondary outcomes included efficacy based on RECIST criteria. An independent Data and Safety Monitoring Board (DSMB) conducted planned safety analyses after the first three patients completed at least 28 days of therapy and, thereafter, to determine dose escalation. Accrual to the study proceeded in accordance with the “3 + 3” study design; accrual was held until the DSMB met and approved escalation to the next highest dose per protocol. The clinical site investigators and independent radiology review both used RECIST criteria to perform tumor assessments at baseline and every 8 weeks thereafter while patients were on the study, even if they had stopped taking BZL101.
Before final analyses of the data, a comprehensive descriptive and exploratory examination was conducted to identify gross errors and potential outliers, and to describe the distribution of the variables in the study sample, both overall and with respect to several key variables. Means, medians, ranges, and standard deviations for continuous variables and frequency distributions for categorical variables were calculated for the entire sample and within sub-samples of interest.
Results
Patient characteristics
Between April 2007 and September 2008, 27 patients were sequentially enrolled to four dose cohorts at eight centers in the United States. As seen in Table 2, the median age was 59 years, 59% were Caucasian and 59% had an ECOG Performance Status of 0. Most patients had received multiple prior treatments, reflecting their advanced disease stage. At baseline, patients had received a median of six (range 1–21) prior anticancer treatments (including chemotherapy, anti-estrogen therapy and targeted therapies) since diagnosis with MBC and a median of two (range 0–10) prior chemotherapy regimens since diagnosis with MBC. As seen in Table 2, there was a median of 2 years (range 0–17 years) since diagnosis of metastatic disease. Eighteen patients (67%) discontinued the study because of disease progression, two patients (7%) discontinued because of SAEs, two patients (7%) discontinued because of adverse events, four patients (15%) discontinued because of patient choice, and one patient (4%) discontinued because of non-compliance with study procedures.
Treatment
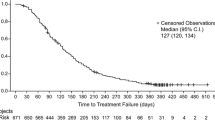
All 27 enrolled patients received BZL101 therapy; 11 patients in cohort 1 (BZL101 10 g/day), six in cohort 2 (BZL101 20 g/day), three in cohort 3 (BZL101 30 g/day), and seven in cohort 4 (BZL101 40 g/day). The median length of drug exposure was 35 days (range 4–449 days). Median treatment compliance was 92% overall (range 61–100%), and 77% of patients took more than 80% of prescribed doses. Overall treatment compliance was calculated as the percentage of the number of prescribed doses taken divided by the number of prescribed doses. No patient underwent a dose reduction (Table 3). The MTD was not reached, thus per protocol, the MTD was defined as the MAD of BZL101, 40 g/day.
Eleven patients were enrolled to the first cohort, BZL101 10 g/day, because when the protocol was originally written, the eligibility criteria did not put a limit on the number of prior chemotherapy regimens allowed for MBC. This meant that patients who were enrolled to the 10 g/day cohort had been treated with multiple prior chemotherapy regimens for MBC (one patient had been treated with 10 prior chemotherapy regimens for MBC), and many patients had disease progression before reaching 28 days on study medication. The DSMB suggested amending the protocol to require patients to have no more than three prior chemotherapy regimens for MBC, which allowed the dose escalation to proceed more effectively in future dose cohorts.
Safety
Twenty-three patients reported adverse events related to BZL101. The primary adverse events associated with BZL101 were grade 1 and 2 gastrointestinal adverse events including: diarrhea, nausea, vomiting, anorexia, distension/bloating, abdominal pain, and flatulence. Drug-related adverse events occurring in more than 5% of patients are displayed in Table 4 with no discernible relationship between grade 3 and 4 adverse events and the dose of BZL101. Three patients reported four DLTs (Table 5). No DLTs were reported in the cohort receiving BZL101 30 g/day.
Ten patients reported 12 SAEs, one of which was classified by the clinical site investigator as related to BZL101: rib pain secondary to vomiting.
Efficacy
Using the available follow-up data, the best overall assessment of response from both the investigator and independent radiology review is shown in Table 6. No patient experienced either a complete or partial response. According to investigator-based assessments, five patients (19%) had stable and 19 (70%) progressive disease. Tumor progression was unknown for three patients (11%). Independent radiology assessment showed that six patients (22%) had stable disease, eight (30%) had progressive disease, and 13 (48%) had unknown response. There were a large number of unknown responses by independent radiology assessment due to many participants having disease progression based on clinical judgment, and thus were not re-staged at study termination. Also, photographs of new skin lesions were not sent for independent review, so independent radiology counted participants with disease progression due to new skin lesions as unknown responders. In addition, according to the independent radiology review, three patients had objective evidence of tumor regression consistent with minimal tumor responses (>0% and <30%).
Fourteen of the 27 patients were on the trial for 28 days or more and were evaluable based on RECIST criteria. Three had stable disease for >120 days (21%). Four patients discontinued BZL101 treatment with stable disease. One patient, who was on BZL101 for 449 days, had objective evidence of minimal tumor regression; she continues with stable disease for 700+ days. A second patient remained stable for 836 days and a third patient remained stable for 594 days; after discontinuing BZL101, both patients did not initiate any further treatment until evidence of progression. The fourth patient was stable for 279 days after discontinuing BZL101.
Discussion
This second phase 1 clinical trial of BZL101 for MBC shows that treatment with a botanical whole drug extract is safe and feasible. In contrast to most early phase studies, we conducted both phase 1 trials in women with MBC rather than in a cohort of patients with tumors of various tissue types because of the selective potency of BZL101 in preclinical breast cancer in vitro and in vivo models. This phase 1B dose escalation trial did not reach a MTD and thus the MTD was defined as the highest dose tested in the study, BZL101 40 g/day. This dose is equivalent in mass to four times the previously evaluated dose in the phase 1A trial.
Unlike the original crude liquid extract administered in the phase 1A trial, the drug in this trial was a freeze-dried powder mixed with sweeteners and taste-enhancing excipients to mask the bitterness of the extract. In the phase 1A trial, eight of 21 patients chose to stop study medication because of the unpalatable taste of the extract. In this phase 1B trial, no patient discontinued the study because of the taste of BZL101. Tolerability of the new formulation was excellent with a median compliance of 92%. The most common adverse events were minor gastrointestinal side effects, with diarrhea the most frequently cited adverse event. The drug product has a large quantity of high molecular weight carbohydrates containing soluble fiber. Further optimization of the purification process to remove the fiber will likely minimize the drug-related diarrhea. Other oral anticancer agents taken for MBC, such as capecitabine and lapatinib, are associated with more severe grade 3 and 4 diarrhea [11] that can necessitate IV hydration and hospitalization, so if a botanical extract can provide similar activity, this would represent a clear advancement in options for patients with MBC.
In this heavily pre-treated population, at least five patients experienced long periods of stable disease, a possible sign of clinical activity. Four patients discontinued BZL101 when their disease was stable. One of these four patients continues to be stable for an extended period of time (700+ days post-BZL101 treatment). In addition, two of these four patients did not initiate any further therapy prior to having objective evidence of progression at 836 and 594 days. In the phase 1A trial, 16 patients were evaluable by RECIST criteria and five of them demonstrated a minimal response; one patient was, 1 mm in tumor size reduction, short of a partial remission, 29% vs. 30% reduction required for a partial remission. If one of the patient’s non-evaluable lesions was included for RECIST analysis she would have met criteria for a partial remission.
On October 31, 2006, the FDA approved the first botanical drug, sinecatechins, a topical treatment for perianal and genital condyloma, through the FDA’s guidelines for botanical drug products that was adopted in June 2004, countering concerns about the approvability of a drug derived from a complex natural mixture where consistency and the inherent chemical variability of plants can lead to challenging quality controls. Today, the FDA has received more than 350 botanical investigational new drug (IND) applications; BZL101 was one of the first botanical INDs issued. In recent years, renewed scientific interest in the identification of potentially useful medicinal compounds from naturally occurring sources has led to the discovery of novel compounds for the treatment of malaria (artemisinin, from the traditional Chinese plant, Artemisia), tyrosinemia (nitrisinone, from the Australian desert plant, Callestimon citrinus), and Alzheimer’s disease [galantamine, isolated from a member of the Amaryllidaceae (Snowdrop) family] [12]. Nearly 30% of all new chemical entities discovered in the last 20 years have been derived from natural products, while a further 20% are chemical adaptations of naturally derived products [13]. More specifically, for drugs approved to treat cancer and infectious diseases, over 60% and 75%, respectively, of all therapies were originally derived from natural products [14].
Most of the prior work on natural products focuses on identifying a single active chemical with significant enhanced activity per extract mass. The current drug development pathway of BZL101 departs from traditional pharmacognosy in that the biological response appears to be dependent on simultaneous cytotoxic activity by a group of compounds rather than by just one. In fact, the active components in BZL101 could be enriched chromatographically but further isolation of the pure compounds resulted in a reduction of activity in in vitro assays compared to the whole extract. This suggested a classic additive or synergistic activity pattern for the components of BZL101; this hypothesis is supported by preliminary results on synergy between phytochemicals purified from the active fractions of BZL101. Nine compounds have been purified from BZL101, all of which belong to the flavonoid family of phytochemicals. Most of these have some cytotoxic activity in vitro, but only at concentrations far higher than those found in the total extract. Although, theoretically, cytotoxicity could be improved by combining different isolated flavonoids, we have not found any combination of the purified actives to be more cytotoxic than the whole extract.
This approach may provide new challenges to the definitions of dose and therapeutic index, now defined by the quantity of a single chemical entity. In the case of BZL101, the FDA defines dose by the total mass of the extract and not by the cumulative mass of the active compounds. Furthermore, conducting pharmacokinetic studies requires more sophisticated tandem methodologies to characterize each active component and their relationships to each other. In future studies, we will attempt to describe the relationship of response with pharmacokinetic profiles of the known active chemical compounds. In addition, since the mechanism for the biological response has been studied and potential clinical response is observed, studies on biomarkers for response, or for patient selection, can be carried out to potentially replace traditional pharmacological analyses aiding clinicians to better understand the therapeutic index of BZL101. It is plausible that this unique mixture will open the opportunity for an alternative approach to cancer therapies, capitalizing on inter-disciplinary analyses of the relationships between the biological response and polychemical drugs.
In summary, this second phase 1 clinical trial demonstrated that BZL101 is safe and well tolerated with encouraging evidence of clinical activity. With an improved formulation of BZL101, a phase 2 clinical trial for women with MBC is planned.
References
Horner MJ, Krapcho M, Neyman N et al (eds) (2009) SEER Cancer Statistics Review: 1975–2006. Bethesda, MD, National Cancer Institute, http://seer.cancer.gov/csr/1975_2006/, based on November 2008 SEER data submission, posted to the SEER web site
Guarneri V, Conte P (2009) Metastatic breast cancer: therapeutic options according to molecular subtypes and prior adjuvant therapy. Oncologist 14(7):645–656
Slamon DJ, Leyland-Jones B, Shak S et al (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344(11):783–792
Campbell MJ, Hamilton B, Shoemaker M, Tagliaferri M, Cohen I, Tripathy D (2002) Antiproliferative activity of Chinese medicinal herbs on breast cancer cells in vitro. Anticancer Res 22(6C):3843–3852
Shoemaker M, Hamilton B, Dairkee SH, Cohen I, Campbell MJ (2005) In vitro anticancer activity of twelve Chinese medicinal herbs. Phytother Res 19(7):649–651
Fong S, Shoemaker M, Cadaoas J et al (2008) Molecular mechanisms underlying selective cytotoxic activity of BZL101, an extract of Scutellaria barbata, towards breast cancer cells. Cancer Biol Ther 7(4):577–586
Shi DY, Xie FZ, Zhai C, Stern JS, Liu Y, Liu SL (2009) The role of cellular oxidative stress in regulating glycolysis energy metabolism in hepatoma cells. Mol Cancer 8:32
Rugo H, Shtivelman E, Perez A et al (2007) Phase I trial and antitumor effects of BZL101 for patients with advanced breast cancer. Breast Cancer Res Treat 105(1):17–28
Le Tourneau C, Lee JJ, Siu LL (2009) Dose escalation methods in phase I cancer clinical trials. J Natl Cancer Inst 101(10):708–720
U.S. Food and Drug Administration: What is a serious adverse event? 8/2009 update. http://www.fda.gov/Safety/MedWatch/HowToReport/ucm053087.htm
Geyer CE, Forster J, Lindquist D et al (2006) Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med 355(26):2733–2743
Butler MS (2004) The role of natural product chemistry in drug discovery. J Nat Prod 67(12):2141–2153
Newman DJ, Cragg GM (2007) Natural products as sources of new drugs over the last 25 years. J Nat Prod 70(3):461–477
Newman DJ, Cragg GM, Snader KM (2003) Natural products as sources of new drugs over the period 1981–2002. J Nat Prod 66(7):1022–1037
Acknowledgments
This work was supported and funded by Bionovo, Inc. located in Emeryville, CA.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Perez, A.T., Arun, B., Tripathy, D. et al. A phase 1B dose escalation trial of Scutellaria barbata (BZL101) for patients with metastatic breast cancer. Breast Cancer Res Treat 120, 111–118 (2010). https://doi.org/10.1007/s10549-009-0678-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-009-0678-5