
Abstract
Endocrine therapy is probably the most important systemic therapy for hormone receptor positive breast cancer. Hormonal manipulation was the first targeted treatment employed in breast cancer therapy even before the role of the estrogen (ER) and progesterone receptors (PR) had been elucidated. Unfortunately, a substantial proportion of patients, despite being ER and/or PR positive, are either primarily resistant to hormone therapies or will develop hormone resistance during the course of their disease. Signaling through complex growth factor receptor pathways, which activate the ER are emerging as important causes of endocrine resistance. Targeted therapies, such as signal transduction inhibitors (STIs), are being explored as agents to be able to potentially overcome this crosstalk and thus, resistance to hormone treatment. This article reviews the biology of the ER, the proposed mechanisms of endocrine resistance, and ongoing clinical trials with STIs in combination with hormonal manipulation as a means to overcome endocrine resistance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The estrogen receptor (ER) is expressed in approximately 70% of patients with breast cancer and plays a critical role in both breast cancer proliferation and metastasis. In spite of high levels of ER expression however, many ER positive patients demonstrate intrinsic resistance to hormonal therapies and all patients with advanced disease eventually develop acquired resistance to endocrine manipulation. It appears that overactive growth factor receptor signaling through multiple intracellular pathways may contribute to the evolution of an endocrine resistance phenotype. Manipulation of growth factor signaling networks has emerged as an attractive strategy delay the onset of, or potentially even overcome, resistance to endocrine therapy in breast cancer.
Search strategy and selection criteria
Data for this review were identified by searches of MEDLINE, Current Contents, PubMed, and references from relevant articles using the search terms “endocrine resistance”, “hormone resistance”, and “estrogen receptor”. In addition, the clinical trials website “clinicaltrials.gov” was searched for all relevant clinical trials. Abstracts and reports from meetings were included only when they related directly to previously published work. Only papers published in English between 1965 and 2006 were included.
Estrogen receptor biology
Estrogen receptor belongs to the steroid–retinoid receptor superfamily, which includes other steroid hormones, retinoic acid, vitamin D, and thyroid-hormone [1]. In the “classical” model of ligand-activated transcription, the steroidal estrogen molecule passively diffuses through the cell membrane and binds to ER located in the nucleus. After binding estrogen, ER dissociates from its chaperone proteins, phosphorylates, and dimerizes with another ER. These activated ER-dimers release co-repressor proteins, such as nuclear receptor co-repressor (NCoR), histone deacetylase 1 (HDAC1), and metastasis-associated antigen 1 (MTA-1) and recruit co-activator proteins, such as amplified in breast cancer-1 (AIB1), nuclear-receptor-coactivator-1 (NCoA-1/SRC1) and the p300 and CBP-associated factor (PCAF) [1, 2]. These activated ER-dimer complexes bind to DNA promoter regions, known as estrogen-response elements (EREs), and regulate transcription of genes involved in proliferation, inhibition of apoptosis, and the promotion of angiogenesis, invasion, and metastasis (Fig. 1) [1].

Nuclear initiated estrogen signaling, classical (left) and non-classical (right). Left: Estrogen (E) binds estrogen receptor (ER), induces dimerization of the protein, and activates DNA binding to estrogen response elements (ERE) in the promoter region of target genes. Coactivator proteins, including AIB1, NCoA-1, and PCAF are recruited to the complex and activate transcription of target genes (classical). Right: Estrogen-bound ER acts as a coactivator, binding to fos and jun at other promoter sites such as AP-1, and recruiting other coactivator proteins to activate the transcription of target genes
Two distinct ERs have been identified, ERα and ERβ, which are located on separate chromosomes and encoded by different genes. ERα and ERβ differ in their tissue distribution and ligand-binding characteristics, which may also explain tissue-selective biological responses to estrogen [3]. ERα is most closely associated with carcinogenesis, as the ratio of ERα:ERβ increases when breast cells become increasingly malignant. It has been speculated that ERβ may act as a tumor suppressor, forming dimer complexes with ERα, thereby inhibiting ERα-driven transcription of genes involved in tumor growth and spread [4]. In addition to its role as a classical transcription factor, ER has recently been found to be involved in other pathways of tumorigenesis. One-third of genes regulated by ER do not contain ERE-promoter sequences. ER may also function as a coactivator by binding key proteins involved in the promoter complex, such as fos and jun, and stabilize the binding of other transcription factors to DNA at alternative response elements such as AP-1. The ER-coactivator complex regulates the expression of a variety of proteins involved in cell proliferation and metastasis, such as insulin-like growth factor 1 (IGF-1), myc, cyclin D1, Bcl-2, and collagenase, in this so-called “non-classical” manner (Fig. 1) [1, 2].
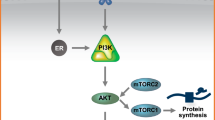
Although the majority of cellular ER is localized to the nucleus, ER has also been associated with the cytoplasm and cell membrane. The ER protein does not contain hydrophobic transmembrane domains or membrane localization sequences. Cellular ER is localized to the cell membrane through its association with cytoplasmic membrane anchors, such as caveolin [5]. There may also be a role for post-translational modifications, alternative splicing variants, and complex formation with alternative transmembrane protein receptors, such as GPR30, to localize ER to the cell membrane [6]. Membrane-bound ER can inappropriately subvert known intracellular signaling pathways to promote cell survival. Membrane-bound ER interacts with members of the type 1 tyrosine kinase growth factor receptor family, such as IGF-1R, EGFR, and HER2. ER associates with Shc at the cytoplasmic membrane surface and induces phosphorylation of IGF-1R leading to phosphoinositide 3-kinase (PI3K) activation and stimulation of the Akt (protein kinase B) pathway [7] PELP1/MNAR is another important coadaptor protein associated with membrane-bound ER. PELP1/MNAR contains phosphoinositide 3-kinase (PI3K) activating domains that can directly activate ERK/MAPK when PELP1/MNAR in associated with ER [8]. PELP1/MNAR may facilitate ER binding to membrane-bound growth factor receptors, such as IGF-1R and EGFR, and induce PI3K-driven activation of Akt. The end result of stimulation of these pathways is increased cell survival, through phosphorylation of pro-apoptotic proteins, such BAD, caspase-9, Forkhead transcription factors and IkappaB kinase alpha [9], and increased transition from G1-S phase in the cell cycle through activation of the downstream effector of Akt, mammalian target of rapamycin (mTOR). Moreover, activated Akt may also phosphorylate nuclear ER and its coregulatory proteins [10], driving a “feed-forward” circuit of ER induced transcriptional activity (Fig. 2) [11].

Membrane-initiated ER signaling. GPR30 and calveolin help localize ER to the cell surface. E2-activated ER associates with PELP/MNAR, which activates membrane-bound growth factor receptors, such as IGF-1R, ErbB2/HER-2, and ErbB1/EGFR, resulting in downstream signaling through Akt and MAPK to activate nuclear-ER and anti-apoptotic proteins, such as BAD, caspase-9, Forkhead transcription factors and IkappaB kinase-α
Thus, there are a variety of means by which the ER can influence cell proliferation and metastasis: through direct DNA binding at ERE-promoter sites, as a coactivator with fos and jun at alternative promoter sites, and through non-genomic molecular “cross-talk” by amplifying proliferative signals through tyrosine receptor kinase growth factor receptors and their downstream effector molecules. Clearly, there exists a complex network of bi-directional cross-talk at multiple levels within breast cancer cells, whereby the ER pathway and growth factor receptor signaling pathways interact and potentiate one another resulting in dysregulated proliferation and growth.
Mechanisms of endocrine-resistance
The goal of hormonal therapy is to block estrogen-induced proliferation of breast cancer cells. Three broad classes of hormonal therapy are used clinically to treat hormone-responsive breast cancer: selective estrogen receptor modifiers (SERMs), such as tamoxifen, which directly bind to the ER and block its transcriptional activity; selective estrogen receptor down-regulators (SERDs), such as fulvestrant, which bind to ER and induce its degradation; and aromatase inhibitors (AIs), such as letrozole, anastrozole, and exemestane, which reduce the production of estrogen through inhibition of aromatase enzyme in peripheral tissues, including bone, muscle and adipose tissue, and within the tumor itself [12].
Although the advent of tamoxifen revolutionized the treatment of breast cancer, almost 50% of ER-positive metastatic breast cancer patients do not respond to tamoxifen in the first-line setting, thereby demonstrating intrinsic resistance. Even if patients respond to tamoxifen initially, they all eventually develop acquired resistance and their disease progresses. Similar findings are seen in the adjuvant setting again confirming the presence of de novo and acquired mechanisms of resistance to tamoxifen. Interestingly though, despite de novo and acquired mechanisms of resistance, many patients who relapse on tamoxifen therapy will respond to further hormonal manipulation with either an AI or fulvestrant, demonstrating that ER continues to play a critical role in proliferation of their breast cancer [13]. Even patients previously treated with tamoxifen and aromatase inhibition were found to have a 19% clinical benefit rate with further hormonal manipulation using fulvestrant in a recent single-centre series [14]. In addition, in the EFECT trial that randomized patients with metastatic breast cancer who had progressed on prior treatment with a non-steroidal aromatase inhibitor to treatment with fulvestrant or exemestane, median duration of benefit was 9.3 and 8.3 months respectively. In this study, >60% of patients had received >2 prior endocrine treatments [15].
Understanding how breast cancer cells develop resistance to hormonal manipulation and finding means of restoring sensitivity to hormonal therapy is a rapidly expanding area in breast cancer research. Some of these mechanisms of “resistance” are in fact due to differences in laboratory tissue processing. For example, significant inter-laboratory variations in the immunohistochemical definition of ER positivity exist. Moreover, there is also intra-laboratory variability as the day of the week when a surgical specimen is received in the laboratory influences ER processing [16]. Loss of ER expression in the evolution of the primary tumor to metastatic disease may contribute to the emergence of estrogen resistance. Data from limited clinical studies suggest that 17% of ER-positive patients treated with adjuvant tamoxifen may convert to an ER-negative phenotype at the time of relapse [17].
Mutations of the ER might also affect the response to hormonal therapy, although studies suggest it occurs in fewer than 10% patients who develop resistance to tamoxifen [18]. Several transcriptional splice variants of ER have been detected in breast cancer specimens that may be involved in endocrine-resistance [19]. For example, ER-βcx, a splice variant of ER-β, is unable to bind estrogen or tamoxifen and induce transcription at EREs. ER-βcx is expressed in 54% of breast cancer specimens as compared with 9% of normal breast tissue [20]. Expression of ER-βcx in breast cancer tissue is associated with lack of expression of PR and resistance to tamoxifen in vitro [21].
There may also be biological variations in tamoxifen metabolizing genes, which influence responsiveness. Tamoxifen is converted to 4-hydroxy-N-desmethyl tamoxifen (endoxifen) through the cytochrome P450 (CYP) 2D6 pathway [22]. Recent studies suggest that levels endoxifen may contribute to the overall anticancer effect derived from tamoxifen and those women with the CYD2D6 *4/*4 genotype have a higher rate of relapse when treated with adjuvant tamoxifen [23, 24].
Beyond genetic modifications of the ER and genetic differences in tamoxifen metabolism, breast cancer cells may develop enhanced sensitivity to estrogen following prolonged hormonal blockade. Numerous in vitro models of long-term estrogen deprivation (LTED) using cell culture techniques with estrogen-depleted media to mimic an oophorectomized state have been published [25, 26]. In response to estrogen deprivation, these cells initially stop growing, but eventually adapt and grow as quickly as wild-type cells cultured in estrogen-rich media. This phenomenon of regrowth has been attributed to the development of adaptive mechanisms of growth in response to residual amounts of estrogen in the culture media. In these models, LTED cells upregulate ER expression and increase localization of ER to the cell membrane. Increased signaling through peptide growth factor pathways, such as IGF-1R and ErbB-2/HER-2, also occurs resulting in MAPK and AKT activation. Stimulation of these signaling pathways, in turn, leads to ligand-independent activation of nuclear ER through phosphorylation, driving transcription of ERE-induced genes involved in proliferation, in spite of low levels of estrogen (Fig. 3).

A model depicting mechanisms of endocrine resistance. Increased growth factor signaling through growth factor receptors, such as IGF-1R, ErbB2/HER-2, and ErbB1/EGFR, leads to ligand-independent activation of nuclear ER through phosphorylation. Growth factor signaling also results increased ER co-activator proteins, such as A1B1 and NCoA-1, which result in TAM-agonist activity on breast cancer cells. Membrane-bound ER can increase growth factor receptor signaling through PELP or directly through Ras/MAPK activation. Increased growth factor signaling may lead to loss of PR expression, and long-term stimulation leads to loss of ER expression and ER-independent growth
More recently, long-term treatment with tamoxifen has been shown to induce a similar estrogen-hypersensitivity phenotype in cell culture, mimicking what may occur in patients who progress after an initial response to tamoxifen [27]. Emerging evidence suggests that tamoxifen may directly induce transcription of epidermal growth factors receptors (EGFR), such as EGFR/HER-1 and ErbB-2/HER-2/neu [28, 29]. In a paired biopsy study of patients who progressed after tamoxifen therapy, increased levels of ErbB-2/HER-2/neu expression and MAPK activity were associated with tamoxifen failure [30]. Thus, long-term treatment with tamoxifen in ER positive breast cancer may increase growth factor receptor-mediated signaling, leading to increased proliferation and escape from hormonal control.
Constitutive activation of peptide-growth factor signaling pathways, particularly the EGFR family, may play a critical role in de novo endocrine resistance. Multiple clinical studies demonstrate that HER-2/neu expression, which occurs in 20–25% of breast cancer, portends a poorer prognosis with tamoxifen therapy [31–33]. It has been suggested that HER-2/neu may confer resistance to tamoxifen by activating ER co-activator proteins, such as A1B1, thereby diminishing the anti-estrogenic effects of tamoxifen and leading to tamoxifen-induced proliferation [34]. Osborne et al. found that co-expression of HER-2/neu and A1B1 conferred a poor prognosis with adjuvant tamoxifen therapy [35]. Interestingly, patients with high HER-2/neu levels alone, without concomitant A1B1 expression, responded to adjuvant tamoxifen as well as patients without HER-2/neu expression. Other studies have suggested that expression of additional ER co-activator proteins, such as NCoA-1/SRC-1, may also mitigate resistance to tamoxifen therapy in HER2/neu-positive tumors [36]. These findings suggest that amplified growth factor signaling, through activation of ER co-activator proteins, may lead to increased tamoxifen agonist activity and tumor growth. The clinical finding that aromatase inhibitors are superior to tamoxifen in HER-2/neu positive patients in the neoadjuvant setting supports this theory that tamoxifen may act as an agonist in breast cancer cells with overactive growth factor receptor signaling networks [37, 38]. It is not yet known if this relative benefit of AI over tamoxifen therapy in HER/neu positive patients exists in the adjuvant setting. A recent retrospective analysis of the BIG 1-98 trial suggested that HER-2/neu overamplification was associated with a high relapse rate, regardless of whether tamoxifen or letrozole was used [39].
Beyond HER-2/neu expression, lack of progesterone receptor (PR) expression may also predict a poorer response to tamoxifen therapy. The expression of PR is regulated by ER activity. For many years, it was theorized that ER+/PR− tumors might not respond as well to hormonal therapy because lack of PR expression reflected a non-functional ER pathway [40]. This hypothesis was supported by the observation that lack of PR expression is associated with decreased response to tamoxifen, shorter time to treatment failure, and poorer overall survival in patients with metastatic disease [41]. Similarly, a meta-analysis of adjuvant tamoxifen therapy for early breast cancer demonstrated that patients with ER+/PR+ tumors benefit more from tamoxifen than patients with ER+/PR− tumors [42]. However, the recent findings of the Arimidex, Tamoxifen, Alone or in Combination (ATAC) trial have challenged traditional theories of hormonal sensitivity of PR negative breast cancers [43]. In the ATAC trial, ER+/PR− patients in the anastrozole alone arm had a similar outcome to ER+/PR+ patients treated with either anastrozole alone, tamoxifen alone, or anastrozole combined with tamoxifen [44]. In contrast, ER+/PR− patients in the tamoxifen arms, either alone or in combination with anastrozole, had a higher recurrence risk than ER+/PR− patients treated with anastrozole alone. Similarly, subgroup analysis of the Intergroup Exemestane Study (IES) also suggested that there may be a differential response AI therapy based upon PR expression, as ER+/PR− showed a non-statistically significant trend toward greater benefit with AI therapy over continued tamoxifen when compared with ER+/PR+ patients [45]. However, the other two large adjuvant AI studies, BIG 1-98 which compared adjuvant letrozole versus tamoxifen and the ABCSG Trial 8/ARNO 95 trial of anastrozole versus tamoxifen after 2 years of adjuvant tamoxifen, have not shown a similar differential benefit for AI therapy for ER+ patients based upon lack of PR expression [46, 47].
Although the role of PR status in predicting response to AI therapy remains controversial, one possible explanation for the differential benefit with AI therapy in PR negative patients in the ATAC trial is overactive growth factor signaling. Loss of PR expression in ER+ breast tumors is associated with increased levels of IGF-1 [48], EGFR/HER-1 [49] and HER-2/neu [50–52] activity. In addition, loss of PR expression is also associated with reduced expression of phosphatase and tensin homolog (PTEN) [53]. PTEN is a critical negative regulator of the PI3K/Akt pathway that transmits downstream intracellular signals from growth factor receptors. IGF-1 is known to downregulate PR expression through Akt-mediated signaling [45]. Studies have shown that high levels of active Akt are associated with an increased risk of relapse for patients treated with adjuvant tamoxifen [54, 55]. Loss of PR expression may also occur with tamoxifen therapy. Sequential biopsy studies have shown that more than half of ER+/PR+ tumors at diagnosis become PR- negative at the time relapse with adjuvant endocrine therapy [56]. One possible explanation is that tamoxifen exerts a selection pressure on ER+/PR+ breast cancer cells causing them to increase growth factor signaling and subsequently lose PR expression with prolonged therapy.
Preclinical data suggests that long-term activation of growth-factor receptor signaling pathways may ultimately lead to an estrogen independent phenotype. While growth-factor receptor signaling initially increases ER transcription and activity, sustained growth-factor signaling activity produces downregulation of ER transcription and activity [57]. For instance, treatment with exogenous EGF, IGF-1, and TGF-β leads to decreased ER-α expression, through increased EGFR, Akt, and protein kinase C (PKC) signaling [58–60]. The events underlying this transition from growth factor driven ER activation to ER downregulation are poorly understood. It has been demonstrated that chronic stimulation of ER by estradiol causes downregulation of ER expression [61]. Sustained growth factor signaling may lead to loss of ER expression in a similar manner and ultimately dissociation from ER-mediated growth. In vitro, breast cancer cells treated with long-term estrogen deprivation upregulate growth-factor signaling activity and evolve into an ER negative phenotype [62, 63].
In summary, there are various mechanisms by which breast cancer cells can escape hormonal control: mutations of ER, increased localization of ER to the cell surface and non-genomic ER activity, expression of splice variants of the ER, upregulation of growth factor receptor expression and intracellular signaling, expression of ER coactivator proteins, and finally, loss of ER expression (Fig. 3). As our understanding of the biology of endocrine therapy improves, the goal of future studies is to prolong response to endocrine manipulation and potentially restore endocrine sensitivity in tumors that have become resistant to endocrine therapy.
Completed and ongoing trials
Extending from research of growth factor signaling in the laboratory setting, a number of clinical trials have sought to determine whether the addition of signal transduction inhibitors to endocrine therapy may overcome endocrine resistance or delay its development. These have included trials using HER2/neu antagonists, tyrosine kinase inhibitors, multikinase inhibitors, and mTOR antagonists.
HER2/neu antagonists
A number of phase II studies have evaluated the combination of trastuzumab and AI therapy in the first and second line setting (Table 1) [64]. The promising results of early combination trials led to the initiation of a phase III trial examining the effectiveness of combination of anastrozole with trastuzumab versus anastrozole alone in women with metastatic ER and/or PR positive, with evidence of HER2neu overamplification [65]. Patients were allowed to have received tamoxifen as adjuvant treatment or as treatment first line for metastatic breast cancer. The primary endpoint was progression-free survival, which was 4.8 months in the combination arm and 2.4 months in the anastrozole alone arm (P = 0.0016). There was also a trend towards improvement in overall survival in the combination arm (28.5 months vs. 23.9 months), which was not statistically significant. The lack of survival benefit in the combination may be explained by the observation that more than 70% of the women in the upfront anastrozole alone arm received trastuzumab upon progression. Of note, there was an increase in the number of grade 3 and 4 adverse events in the combination arm (25% of patients vs. 15% of patients).
Since trastuzumab may not inhibit cells that express low levels of HER2, an alternative strategy using lapatanib, an oral tyrosine kinase inhibitor that inhibits both Erb1 (EGFR) and Erb2 (HER2), is under evaluation. A phase III clinical trial with lapatanib [NCT00073528] in combination with letrozole was launched in 2003 to investigate the combination of letrozole plus lapatanib versus letrozole plus placebo in women with tamoxifen resistant advanced breast cancer. Interestingly, Erb1 and Erb2 positivity is not an inclusion criteria in this trial, although the results will be analyzed to determine the impact of Erb1/Erb2 levels on efficacy of treatment. In addition, an NCI phase II trial [NCT00118157] is currently underway to investigate the efficacy of the combination of tamoxifen and lapatinib in women with tamoxifen resistant locally advanced or metastatic breast cancer. Studies in the future may investigate the role of pertuzumab, an antibody that distrupts Her2 dimerization, which has been shown in vitro to promote the antitumor efficacy of fulvestrant [66].
Tyrosine Kinase inhibitors
A third strategy that is under study is the use of tyrosine kinase inhibitors in combination with endocrine therapy. A phase II double blind multicentre three-arm preoperative window trial of anastrozole alone versus anastrozole plus gefitinib (an oral EGFR inhibitor) in two different combinations (anastrozole plus gefitinib for 16 weeks or anastrozole alone for 2 weeks then anastrozole plus gefitinib for 16 weeks) was conducted in 206 patients, with the primary endpoint being comparison of ki67 (a marker of cell proliferation) at 16 weeks [67]. In this study, there was no significant change in ki67 in these groups, and indeed, a trend against the combination was suggested.
A second study looked at 53 previously untreated postmenopausal patients with ER positive EGFR positive primary breast cancer with randomization to 4 weeks of gefitinib alone versus gefitinib plus anastrozole in a controlled double blind trial. The combination of gefitinib plus anastrozole was more effective than gefitinib alone in reducing Ki67 (−77.3% versus −93.2% P = 0.008) but did not significantly reduce tumor size (−30.3% versus −28.8% P = ns) [68]. In a third trial, 13 postmenopausal patients with resectable, histologically diagnosed hormone sensitive breast cancer were treated with three months of letrozole and imatinib (a PDGFR/C-kit/stem cell factor tyrosine kinase inhibitor) (400 mg twice daily) prior to surgery. Five patients experienced grade 3 toxicity, and three patients were withdrawn from the study after 2 months of treatment. Of 10 evaluable patients, 9 had a clinical partial response and 1 had stable disease. This study concluded that the combination of imatinib and letrozole may be promising however due to toxicity, the dose of imatinib must be reduced [69]. Finally, preliminary analysis of 18 patients in a phase II trial treated with letrozole and erlotinib (an EGFR tyrosine kinase inhibtior) as first or second line hormonal therapy showed clinical benefit (stable disease + partial response + complete response) in 11 patients with time to progression of 13 months. In molecular studies, EGFR expression (by IHC DAKO technique) did not correlate with clinical response [70].
There are multiple ongoing trials (Table 2) exploring the EGFR inhibitors gefitinib and erlotinib in combination with AIs, SERMs (tamoxifen), and SERDs (fulvestrant).
Multikinase inhibitors
At present, it is not known which of the many identified growth factor pathways are the most important in contributing to endocrine resistance. Multikinase inhibitors, which target several intracellular pathways, are therefore being explored in combination with endocrine therapy. One approach has been to use farnesyl transferase inhibitors, which modulate the activity of many classes of intracellular proteins. In a randomized phase II trial, 121 postmenopausal patients with ER positive advanced breast cancer that had progressed after tamoxifen were randomized to letrozole plus placebo versus letrozole plus tipifarnib. While the study was not powered to detect an impact on disease progression, the objective response rate was higher with letrozole alone than in the combination group (38% letrozole group, 26% combination group) [71]. A second phase II trial reported preliminary results in 19 postmenopausal tamoxifen-resistant patients treated with tamoxifen and tipifarnib. In this small group there was one partial response [72]. There is an ongoing phase II trial, which randomizes women with hormone sensitive disease to anastrozole plus lonafarnib or anastrozole alone [NCT 00081510].
Other multikinase inhibitors currently being investigated include sorafenib (which targets receptor tyrosine kinases and serine/threonine kinases) [NCT 00217399] and PTK787/ZK222584 (a VEGFR and TK inhibitor) [NCT 00263198].
VEGFR modulators
Following the introduction of VEGF receptor antagonists in the treatment of other cancers, combining endocrine modulators with VEGF receptor modulators is an attractive approach. A phase I/II trial of letrozole with bevacizumab showed that the combination is well tolerated [73]. Phase II trials are currently recruiting that will investigate whether bevacizumab can restore sensitivity to hormonal manipulation in women who have progressed on endocrine therapy [NCT00187694, NCT00240071].
Rapamycin analogues
The phosphatidylinositol-3-kinase (PI3K) pathway is another signaling pathway that becomes overactive in breast cancer cells, and may lead to endocrine resistance. Molecular target of rapamycin (mTOR) is a downstream key target protein for PI3K. The mTOR inhibitors CCI779 and RAD001 have been examined for their efficacy in combination with endocrine agents. A randomized phase II study of CCI-779 (temsirolimus) in combination with letrozole compared to letrozole alone in patients with advanced breast cancer suggested that progression free survival may be extended in the combination arm [74]. The phase III trial (n = 112) however was stopped early at the recommendation of the Independent Data Monitoring Committee since a greater percentage of patients treated with the combination of temsirolimus and letrozole experienced more grade three toxicity when compared with those treated with letrozole alone (hyperglycemia 4% vs. 1%, neutropenia 3% vs. 1%, asthenia 3% vs. 2%). As well, the combination did not improve progression free survival over treatment with letrozole alone [75]. Studies are ongoing evaluating the value of adding everolimus (RAD001) to letrozole as preoperative therapy of primary breast cancer [NCT 0010116]. In preclinical studies, RAD001 treatment of endocrine-sensitive and endocrine-resistant breast cancer cell lines resulted in a dose-dependant decrease in proliferation and estrogen receptor alpha mediated transcription [76].
Discussion and future research directions
Treatment of endocrine resistant breast cancer is a fascinating and rapidly growing field. The use of signal transduction inhibitors in this setting is still in its infancy. Since we now understand that there are many potential mechanisms of resistance, it makes intuitive sense that resistance to endocrine manipulation will not be the same in every patient. Further elucidation of clinically relevant pathways of resistance, biomarkers reflecting the activity of these pathways and surrogate biomarkers of response are therefore critical to determine which therapeutic strategies should be tested. Recent studies suggest that overexpression of p53 and loss of p27kip1 expression may predict resistance to endocrine therapy [77, 78]. Studies in the neoadjuvant setting may be able to further this research, by allowing for sequential tumor biopsies in order to determine the molecular effects of combining STIs with hormonal treatment. However, caution must be used, particularly in the curative setting, since STIs may potentially accelerate growth and metastasis. For instance, in one study examining the use of gefitinib alone in patients with operable breast cancer, women with ER positive and PR negative and her2 amplified tumors were more likely to show molecular growth inhibition, whilst ER+/PR+ tumors showed molecular growth proliferation [79].
For targeted approaches, patient selection is crucial, as we have learned from the studies of the role EGFR mutations to predict responsiveness to Erlotinib [80] and gene array studies to predicting the outcome with adjuvant chemotherapy [81]. Many of the ongoing studies of STIs in combination with endocrine treatment indeed incorporate molecular profiling in order to be able to predict who will respond to combination treatment. Finally, it is not yet known whether the addition of STIs prior to the development of endocrine resistance may be able to delay the development of hormone resistant breast cancer.
At present, there is a sequence of endocrine treatments that are used in treating ER and/or PR+ advanced breast cancer, and once endocrine options have been exhausted the treatment of choice is chemotherapy. In the future, however, we may be able to intervene and reverse resistance with a sequence of targeted signal transduction inhibitors, thereby delaying the use of chemotherapy in patients with advanced breast cancer.
References
Osborne CK, Schiff R (2005) Estrogen-receptor biology: continuing progress and therapeutic implications. J Clin Oncol 23:1616–1622
Normanno N, Di Maio M, De Maio E et al (2005) Mechanisms of endocrine resistance and novel therapeutic strategies in breast cancer. Endocr Relat Cancer 12:721–747
Yager JD, Davidson NE (2006) Estrogen carcinogenesis in breast cancer. N Engl J Med 354:270–282
Speirs V (1997) Oestrogen receptor in breast cancer: good, bad or still too early to tell? J Pathol 197:143–147
Jacob J, Sebastian KS, Devassy S et al (2006) Membrane estrogen receptors: genomic actions and post transcriptional regulation. Mol Cell Endocrinol 246:34–41
Filardo EJ, Thomas P (2005) GPR30: a seven-transmembrane-spanning estrogen receptor that triggers estrogen release. Trends Endocrinol Metab 16:362–367
Kahlert S, Nuedling S, van Eickels M et al (2000) Estrogen receptor alpha rapidly activates the IGF-1 receptor pathway. J Biol Chem 16:18447–18453
Barletta F, Wong CW, McNally C et al (2004) Characterization of the interactions of estrogen receptor and MNAR in the activation of Src. Mol Endocrinol 18:1096–1108
Johnston SR, Head J, Pancholi S et al (2003) Integration of signal transduction inhibitors with endocrine therapy: an approach to overcoming hormone resistance in breast cancer. Clin Cancer Res Jan(9): 524s–533s
Font de Mora J, Brown M (2000) AIB1 is a conduit for kinase mediated growth factor signaling to estrogen receptor. Mol Cell Biol 20:5041–5047
Simoncini T, Hafezi-Moghadam A, Brazil DP et al (2000) Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 407(6803):538–541
Pietras RJ (2006) Biologic basis of sequential and combination therapies for hormone-resistant breast cancer. Oncologist 11:704–717
Ring A, Dowsett M (2004) Mechanisms of tamoxifen resistance. Endo Relat Cancer 11:643–658
Steger GG, Bartsch R, Wenzel C et al (2005) Fulvestrant (‘Faslodex’) in pre-treated patients with advanced breast cancer: a single-centre experience. Eur J Cancer 41(17):2655–2661
Gradishar W, Chia S, Piccart M, on behalf of the EFECT writing committee (2006) Fulvestrant versus exemestane following prior non-steroidal aromatase inhibitor therapy: first results from EFECT, a randomized, phase III trial in postmenopausal women with advanced breast cancer SABCS 2006: Abstr 12
Nkoy FL, Hammond E, Rees W et al (2005) Day of surgery affects estrogen receptor test results in women with breast cancer SABCS 2005: Abst 5107
Kuukasjarvi T, Kononen J, Helin H et al (1996) Loss of estrogen receptor in recurrent breast cancer is associated with poor response to endocrine therapy J Clin Oncol 14:2584–2589
Achuthan R, Bell SM, Roberts P et al (2001) Genetic events during the transformation of a tamoxifen sensitive human breast cancer cell line into a drug resistant clone. Cancer Genet Cytogenet 130:166–172
Hernyk MH, Fuqua SA (2004) Endocr Rev 25(6):869–898
Omoto Y, Kobayashi S, Inoue S et al (2002) Evaluation of oestrogen receptor ß wild-type and variant protein expression, and relationship with clinicopathological factors in breast cancers. Eur J Cancer 38:380–386
Saji S, Omoto Y, Shimizu C et al (2002) Clinical impact of assay of estrogen receptor β cx in breast cancer. Breast Cancer 9:303–307
Johnson MD, Zuo H, Lee KH et al (2004) Pharmacological characterization of 4-hydroxy-N-desmethyl tamoxifen, a novel active metabolite of tamoxifen. Breast Cancer Res Treat 85:151–159
Goetz MP, Rae JM, Suman VJ et al (2005) Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol 23:9312–9318
Bonanni B, Macis D, Maisonneuve P et al (2006) Polymorphism in the CYP2D6 tamoxifen-metabolizing gene influences clinical effect but not hot flashes: data from the Italian tamoxifen trial. J Clin Oncol 24:3708–3709
Santen RJ, Song RX, Zhang Z et al (2004) Adaptive hypersensitivity to estrogen: mechanism for sequential responses to hormonal therapy in breast cancer Clin Cancer Res 10:337s–345s
Johnston SR, Dowsett M (2002) Aromatase inhibitors for breast cancer: lessons from the laboratory. Nat Rev Cancer 3:821–883
Berstein LM, Wang J-P, Zheng H et al (2004) Long-term exposure to tamoxifen induces hypersensitivity to estradiol. Clin Cancer Res 10:1530–1534
Yarden RI, Wilson MA, Chrysogelos SA (2001) Estrogen of EGFR expression in breast cancer cells: a possible mechanism to modulate growth. J Cell Biochem 81:232–246
Salvatori L, Pallante P, Ravenna L et al (2003) Oestrogens and selection estrogen receptor (ER) modulators regulate EGF receptor gene expression through ER-alpha and beta subtypes via an SP-1 site. Oncogene 22:4875–4881
Gutierrez MC, Detre S, Johnston S et al (2005) Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J Clin Oncol 23:2469–2476
Mass R (2000) The role of HER-2 expression in predicting response to therapy in breast cancer. Semin Oncol 27:46–52
Ciocca DR, Elledge R (2000) Molecular markers for predicting response to tamoxifen in breast cancer patients. Endocrine 13:1–10
De Laurentiis M, Arpino G, Massarelli E et al (2005) A meta-analysis on the interaction between HER-2 expression and response to endocrine treatment in advanced breast cancer. Clin Cancer Res 11:4741–4748
Font de Mora J, Brown M (2000) A1B1 is a conduit for kinase-mediated growth factor signaling to the estrogen receptor. Mol Cell Biol 20:5041–5047
Osborne CK, Bardou V, Hopp TA et al (2003) Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst 95(5):353–361
Fleming FJ, Myers E, Kelly G et al (2004) Expression of SRC-1, AIB1, and PEA3 in HER2 mediated endocrine resistant breast cancer; a predictive role for SRC-1. J Clin Pathol 57:1069–1074
Ellis MJ, Coop A, Singh B et al (2001) Letrozole is more effective neoadjuvant endocrine therapy than tamoxifen for ErbB-1- and/or ErbB-2-positive, estrogen receptor-positive primary breast cancer: evidence from a phase III randomized trial. J Clin Oncol 19:3808–3816
Smith IE, Dowsett M, Ebbs SR et al (2005) Neoadjuvant treatment of postmenopausal breast cancer with anastrozole, tamoxifen, or both in combination: the immediate preoperative anastrozole, tamoxifen, or combined with tamoxifen (IMPACT) multicenter double-blind randomized trial. J Clin Oncol 23:5108–5116
Mauriac L, Keshaviah A, Debled M et al (2007) Predictors of early relapse in postmenopausal women with hormone receptor-positive breast cancer in the BIG 1-98 trial. Ann Oncology (advanced e-publishing)
McGuire WL, Horwitz KB, Pearson OH et al (1977) Current status of estrogen and progesterone receptors in breast cancer. Cancer 39:2934–2947
Ravdin PM, Green S, Dorr TM et al (1992) Prognostic significance of progesterone receptor levels in estrogen receptor-positive patients with metastatic breast cancer treated with tamoxifen: results of a prospective Southwest Oncology Group study. J Clin Oncol 10:1284–1291
Bardou VJ, Arpino G, Elledge RM et al (2003) Progesterone receptor status significantly improves outcome prediction over estrogen receptor status alone for adjuvant endocrine therapy in two large breast cancer databases. J Clin Oncol 21:1973–1979
Cui X, Schiff R, Arpino G et al (2005) Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy. J Clin Oncol 23:7721–7735
Dowsett M, Cuzick J, Wale C et al (2005) Retrospective analysis of time to recurrence in the ATAC trial according to hormone receptor status: an hypothesis-generating study. J Clin Oncol 23:7512–7517
Coombes RC, Hall E, Gibson LJ et al (2004) A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer. N Engl J Med 350:1081–1092
Thürlimann B, Keshaviah A, Coates AS et al (2005) A comparison of letrozole and tamoxifen in postmenopausal women with early breast cancer. N Engl J Med 353:2747–2757
Jakesz R, Jonat W, Gnant M et al (2005) Switching of postmenopausal women with endocrine-responsive early breast cancer to anastrozole after 2 years’ adjuvant tamoxifen: combined results of ABCSG trial 8 and ARNO 95 trial. Lancet 366:455–462
Cui X, Zhang P, Deng W et al (2003) Insulin-like growth factor-I inhibits progesterone receptor expression in breast cancer cells via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin pathway: progesterone receptor as a potential indicator of growth factor activity in breast cancer Mol Endocrinol 17:575–588
Arpino G, Weiss H, Lee AV et al (2005) Estrogen receptor-positive, progesterone receptor negative breast cancer: association with growth factor receptor expression and tamoxifen resistance. J Natl Cancer Inst 97:1254–1261
Balleine RL, Earl MJ, Greenberg ML et al (1999) Absence of progesterone receptor associated with secondary breast cancer in postmenopausal women. Br J Cancer 79:1564–1571
Bamberger AM, Milde-Langosch K, Schulte HM et al (2000) Progesterone receptor isoforms, PR-B and PR-A, in breast cancer: correlations with clinicopathologic tumor parameters and expression of AP-1 factors. Horm Res 54:32–37
Dowsett M, Harper-Wynne C, Boeddinghaus I et al (2001) HER-2 amplification impedes the antiproliferative effects of hormone therapy in estrogen receptor-positive primary breast cancer. Cancer Res 61:8452–8458
Shi W, Zhang X, Pintilie M et al (2003) Dysregulated PTEN-PKB and negative receptor status in human breast cancer. Int J Cancer 104:195–203
Kirkegaard T, Witton CJ, McGlynn LM et al (2005) AKT activation predicts outcome in breast cancer patients treated with tamoxifen. J Pathol 207:139–146
Perez-Tenorio G, Stal O (2002) Activation of AKT/PKB in breast cancer predicts a worse outcome among endocrine treated patients. Br J Cancer 86:540–545
Gross GE, Clark GM, Chamness GC et al (1984) Multiple progesterone receptor assays in human breast cancer. Cancer Res 44:836–840
Nicholson RI, Staka C, Boyns F et al (2004) Growth-factor driven mechanisms associated with resistance to estrogen deprivation in breast cancer: new opportunities for therapy. Endo-Relat Cancer 11:623–641
Stoica A, Saceda M, Fakhro A, Salomon HB et al (1997) The role of transforming growth factor-beta in regulation of estrogen receptor expression in the MCF-7 breast cancer cell line. Endocrinology 138:1498–1505
Stoica A, Saceda M, Fakhro A et al (2000) Role of insulin-like growth factor-I in regulating estrogen receptor-alpha gene expression. J Cell Biochem 76:605–614
Stoica A, Saceda M, Doraiswamy VL et al (2000) Regulation of estrogen receptor-alpha gene expression by epidermal growth factor. J Endocrinol 165:371–378
Pink JJ, Jordan VC (1996) Models of estrogen receptor regulation by estrogens and antiestrogens in breast cancer cell lines. Cancer Res 56:2321–2330
Murphy CS, Pink JJ, Jordan VC (1990) Characterization of a receptor-negative, hormone-nonresponsive clone derived from a T47D human breast cancer cell line kept under estrogen-free conditions. Cancer Res 50:7285–7292
Pink JJ, Bilimoria MM, Assikis J et al (1996) Irreversible loss of the estrogen receptor in T47D breast cancer cells following prolonged estrogen deprivation. Br J Cancer 74:1227–1236
Marcom PK, Isaacs C, Harris L et al (2005) A phase II trial of letrozole and trastuzumab for ER and/or PgR and Her2 positive metastatic breast cancer: final results. J Clin Oncol 23:596
Kaufmann B, Mackey J, Clemons M et al (2006) Trastuzumab plus anastrozole prolongs progression-free survival in postmenopausal women with her2-positive, harmone-dependent metastatic breast cancer (MBC). Ann Oncol 17(Suppl 9): LBA2
Pietras RJ, Marquez DC, Chen H et al (2006) Improved antitumour therapy by dual targeting of estrogen and growth factor receptor signaling in human breast cancer cells. J Clin Oncol 24(18 Suppl):637
Dowsett M, Smith I, Skene A et al (2006) (Study 0223) Biological and clinical outcomes from a phase II placebo controlled neoadjuvant study of anastrozole alone or with gefitinib in postmenopausal women with ER/PGR+ breast cancer. J Clin Oncol 24(18 Suppl):515
Polychronis A, Sinnett HD, Hadjiminas D et al (2006) Anti-proliferative and molecular effects of neoadjuvant (pre-operative) gefitinib alone or in combination with anastrozole in epidermal growth factor receptor (EGFR) positive, estrogen receptor alpha (ERa) positive patients with primary breast cancer. J Clin Oncol 23(16 Suppl):552
Chow LW, Yip AY, Toi M (2006) Evaluation of neoadjuvant inhibition of aromatase activity and signal transduction in breast cancer Ann Oncol 17(Suppl 9): Abs422P
Mayer IA, Granja NM, Shyr Y et al (2006) A phase II trial of letrozole plus erlotinib in postmenopausal women with hormone sensitive matestatic breast cancer: preliminary results of toxicities and correlative studies. SABCS 2006: Abs4052
Johnson SRD, Semiglazov V, Manikhav G et al (2005) A randomized, blinded, phase II study of tipifarnib (Zarnestra) combined with letrozole in the treatment of advanced breast cancer after antiestrogen therapy. SABCS 2005: Abstr 5087
Dolenc F, Lacroix-Tikri M, Mourey L et al (2005) Tipifarnib with tamoxifen as a rescue for tamoxifen acquired clinical resistance for metastatic ER and/or PgR positive breast cancer after relapse under tamoxifen. Preliminary results SABCS 2005: Abstr 5098
Traina TA, Rugo H, Caravelli J et al (2006) Letrozole with bevacizumab is feasible in patients with hormone receptor positive metastatic breast cancer. J Clin Oncol 24(18 Suppl):3050
Carpenter JT, Roche H, Campone M et al (2005) Randomized 3-arm, phase 2 study of temsirolimus (CCI-779) in combination with letrozole in postmenopausal women with locally advanced or metastatic breast cancer. J Clin Oncol 23(16 Suppl):564
Chow LWC, Sun Y, Jassem J et al (2006) Phase III study of temsirolimus with letrozole or letrozole alone in postmenopausal women with locally advanced or metastatic breast cancer SABCS 2006: Abstr 6091
Farmer I, Evans DB, Lane HA et al (2006) Preclinical studies of the combination of RAD001 with tamoxifen or letrozole in breast cancer. Eur J Cancer Supplements 4(2):142: Abstr 324
Kai K, Nishimura R, Matsuda M et al (2005) P53 overexpression is a significant factor in predicting resistance to 3rd generation aromatase inhibitors (AIs) in hormone-sensitive recurrent or advanced breast cancer. J Clin Oncol 23(16 Suppl):715
Ellis MJ, Too Y, Lin L et al (2006) High nuclear (N) p27kip1 levels predict sensitivity to neoadjuvant letrozole in ER+ breast cancer independent of AKT levels, PIK3CA mutation status, and cell cycle effects: a potential role for p27kip1 in predicting enhanced autophagocytosis in response to aromatase inhibitor therapy. SABCS 2006: Abstr 4048
Finn RS, Dering J, Ginther C et al (2006) ER+ PR- breast cancer defines a unique subtype of breast cancer that is driven by growth factor signaling and may be more likely to respond to EGFR targeted therapies. J Clin Oncol 24(18 Suppl):514
Sakurada A, Shepherd FA, Tsao M-S (2006) Epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer: impact of primary or secondary mutations. Clin Lung Cancer 7(Suppl 4):S138–S144
Hanneman J, Oosterkamp HM, Bosch CAJ et al (2006) Changes in gene expression associated with response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol 23:3331–3342
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bedard, P.L., Freedman, O.C., Howell, A. et al. Overcoming endocrine resistance in breast cancer—are signal transduction inhibitors the answer?. Breast Cancer Res Treat 108, 307–317 (2008). https://doi.org/10.1007/s10549-007-9606-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-007-9606-8