
Abstract
Tyrosinemia type I is a recessive inborn error of metabolism caused by mutations in the fumarylacetoacetate hydrolase (FAH) gene, coding for the final enzyme in the metabolism of tyrosine. This renders FAH nonfunctional and without treatment, toxic metabolites accumulate causing liver and kidney damage. Introduction of the drug NTBC in 2002 offered a treatment which inhibits an upstream enzyme, preventing the production of the toxic metabolites. There is now a long-term survival rate of greater than 90 % in children, but there are reports of lower cognitive function and IQ as well as schooling and behavioral problems in these children. We studied a mouse model of tyrosinemia type I to gain insight into the effects of tyrosinemia type I and treatment with NTBC on mouse learning, memory, and behavior. In the Barnes maze, visual and spatial cues can be used by mice to remember the location of a dark escape box. The primary time, distance, and strategy taken by the mice to locate the escape box is a measure of learning and memory. Our findings show that mice with tyrosinemia type I were slower to learn than wild-type mice treated with NTBC and made more mistakes, but were capable of learning and storing long-term memory. After learning the location of the target hole, mice with tyrosinemia type I respond differently to a change in location and were less flexible in learning the new target hole location. Our findings suggest that this slower learning and cognitive difference is caused by tyrosinemia type I and not by the treatment with NTBC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
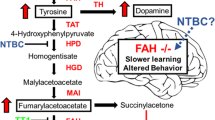
Tyrosinemia type I (TT1, OMIM 276700) is a recessive inborn error of metabolism caused by mutations in the fumarylacetoacetate hydrolase gene (FAH, EC 3.7.1.2). This enzyme catalyzes the fourth and final step in the breakdown of tyrosine (Fig. 1). The reduced activity or absence of expression of the FAH enzyme results in the accumulation of the metabolites proximal to this step, including malylacetoacetate (MAA) and fumarylacetoacetate (FAA), both of which can be converted to succinylacetone (SA). This causes severe liver and kidney damage (Lindblad et al 1977). FAA has been shown to be a mutagenic metabolite and a major risk factor for hepatocellular carcinoma (Jorquera and Tanguay 2001). Succinylacetone is a neurotoxin and can interact with renal transport proteins causing a tubular wasting and Fanconi syndrome with secondary rickets (Santra et al 2008; Maiorana et al 2014). Succinylacetone is also a very potent inhibitor of ALAD, the rate limiting enzyme in heme synthesis, causing a build-up of δ-ALA (5-aminolevulinic acid) and porphyria-like peripheral neuropathy symptoms and CNS neurological crisis. The diagnosis of tyrosinemia type I is made by the detection of succinylacetone in the plasma or urine (Lindstedt et al 1992; Barnby 2014) and is confirmed by finding mutations in the FAH gene.

The tyrosine catabolism and dopamine synthesis pathways. Tyrosine can be ingested from food, or synthesized by the hydroxylation of phenylalanine by phenylalanine hydroxylase (PAH, EC 1.14.16.1). The first step in the breakdown of tyrosine is conversion to 4-hydroxyphenylpyruvate by tyrosine aminotransferase (TAT, EC 2.6.1.5). The second step is conversion into homogentisate, by 4-hydroxyphenylpyruvate dioxygenase (HPPD, EC 1.13.11.27) and this is the step that is inhibited by the pharmaceutical 2-[2-nitro-4-trifluoromethylbenzoyl) cyclohexane-1,3-dione (NTBC). NTBC prevents the breakdown of tyrosine causing hypertyrosinemia. Homogentisate is oxidized to malylacetoacetate by homogentisate dioxygenase (HGD, EC 1.13.11.5) and isomerized by maleylacetoacetate isomerase (MAI, EC 5.2.1.2) to form fumarylacetoacetate. The final step of tyrosine metabolism involves the breakdown of fumarylacetoacetate into fumarate and acetoacetate, and is catalyzed by fumarylacetoacetate (FAH, 3.7.1.2). This is the enzyme mutated in tyrosinemia type I. Tyrosine serves as the basic building block for dopamine. First tyrosine is hydroxylated to L-DOPA by tyrosine hydroxylase (TH, EC 1.14.16.2), and then dopamine is synthesized by decarboxylation of L-DOPA by DOPA decarboxylase (DDC, EC 4.1.1.28). There are several pathways by which dopamine can be broken down but they involve oxidation by monoamine oxidase (MAO, EC 1.4.3.4) and modification by catechol-O-methyltransferase (COMT, EC 2.1.1.6) to produce the stable waste product homovanillate. In the case of tyrosinemia type I (TTI) where the enzyme FAH is compromised, MAA and FAA will accumulate and be converted to the metabolite succinylacetone which has deleterious effects on the liver, kidneys, and the central and peripheral nervous system. The rate-limiting enzyme in heme synthesis δ-aminolevulinate dehydratase (ALAD, EC 4.2.1.24) is extremely sensitive to inhibition by SA, leading to a buildup of δ-aminolevulinate (δ-ALA) and subsequent toxic effects
Although most of the long term complications from the disease come from hepatic failure and hepatocellular carcinoma, there are frequent episodes of peripheral neuropathy and neurological crisis. This is characterized by excruciating pain and peripheral muscle weakness (Mitchell et al 1990) that can be completely reversed after liver transplantation (Noble-Jamieson et al 1994). Around 10 % of deaths have occurred during neurological crisis (Van Spronsen et al 1994). For years the only treatment for children with tyrosinemia type I was a liver transplant and/or a diet low in tyrosine and phenylalanine. Since the approval of NTBC (2-(2-nitro-4-trifluoromethylbenzoyl) cyclohexane-1-3-dione) for treatment of tyrosinemia type I in 2002 (in the US), it has been established as a highly-effective treatment and an alternative to liver transplantation. Before treatment with NTBC there was a high mortality before two years of age, usually from hepatocellular carcinoma and/or hepatic encephalopathy. Children in the US have been treated with NTBC for over a decade (longer in Europe), and follow up examinations are showing lower than normal IQ, schooling difficulties, as well as behavioral, cognitive, and learning problems (Masurel-Paulet et al 2008; De Laet et al 2011; Thimm et al 2012; Pohorecka et al 2012; Bendadi et al 2014). Children with tyrosinemia type I being treated with NTBC had an average IQ score 20 points lower than their unaffected siblings (Bendadi et al 2014). Children with tyrosinemia type I also show altered cognitive, learning, schooling, and behavioral problems. The mechanism(s) behind these changes are unknown. Proposed mechanisms include hypertyrosinemia and increased transport of tyrosine into the brain, decreased transport of neutral amino acids into the brain, increased CNS dopamine, decreased CNS serotonin, oxidative damage from δ-ALA, and succinylacetone modification of neuronal proteins. Succinylacetone has been shown to accumulate in the rat brain and be a potent inhibitor of heme synthesis (Wyss et al 1993).
Years before the introduction of NTBC treatment, it was established that there was brain pathology associated with tyrosinemia type I, especially of the white matter of the brain (Kobayashi et al 1980). There have been autopsy findings of spongy degeneration of the white matter in the central cerebrum (Partington and Haust 1967) and multiple reports of alterations in astroglial cells with large, vacular nuclei in some cases and poorly developed white matter in another case (Halvorsen et al 1966). White matter composes about half of the human brain and hypomyelination has been implicated in altered cognitive function. Increases in cortical myelination occur during development and skill learning (Fields 2008). Hypomyelination or dysmyelination cause nerve conduction impairment resulting in cognitive changes and behavioral disorders (Fields 2008).
It remains uncertain whether the cognitive and behavioral problems observed in children with tyrosinemia type I are a long term consequence of tyrosinemia type I, or are caused by treatment with NTBC. To gain insight into this question we used the Barnes maze to test learning and memory in mice with and without tyrosinemia type I treated with NTBC (Barnes 1979).
Materials and methods
Tyrosinemia type I mice
This Fah5961SB (Fah1R or Fahmut) genetically modified mouse was originally created at Oak Ridge National Laboratories (Aponte et al 2001). The mutation generated is a G-to-A substitution at the last base of exon 7 leading to the splicing of exon 6 to exon 8, and resulting in a transcript that lacks exon 7. The absence of exon 7 in the transcript results in a frameshift and subsequently the introduction of a premature stop codon at amino acid position 303 (Aponte et al 2001). This results in no functional FAH protein expressed in the mouse. We obtained the Fah1RTyrC/RJ mice (Stock# 018129) from Jackson Labs (Bar Harbor, Maine, USA). For additional details about the tyrosinemia type I mice, their genotyping, urine analysis, and nesting building behavior (Deacon 2006), see Supplementary materials.
Each experimental cohort consisted of three groups, WT mice drinking water, WT mice drinking NTBC dissolved in water (7.5 mg/L), and tyrosinemia type I mice Fahmut/Fahmut drinking NTBC dissolved in water (7.5 mg/L). The pregnant dams are treated with NTBC in their drinking water a few days after mating. The Fahmut/Fahmut experimental pups will die within 24 hrs if they do not receive NTBC from their mother’s breast milk. From now on we will refer to the Fah5961SB/Fah5961SB mice as Fahmut. We tested two cohorts of mice from time separated breeding’s with a final total of 8–10 animals per group. Mice were weaned and separated at four weeks and behavioral testing was conducted when the mice were between 12 and 16 weeks of age.
Mice were housed on a 12 hr reversed light/dark cycle, housed 2-5 mice per cage, with corn cob bedding (Harlan), fed mouse chow (Teklad Global 18 % Protein Rodent Diet which contained 0.6 % tyrosine and 1 % phenylalanine), and water ad libitum. NTBC was purchased from Yecuris Corporation (Tualatin, OR). Mouse houses and wheels as well as cotton nesting material were provided for enrichment purposes. There was no difference in mouse weight between the three groups of mice (male mice only at 12-14 weeks) F (2, 29) = 0.6062, P = 0.5522, with WT-water mice weighing 27.8 ± 0.1 g (n = 8) and WT-NTBC and Fahmut-NTBC mice weighing 26.4 ± 0.7 g (n = 10) and 26.9 ± 0.8 g (n = 14), respectively. All mouse care, breeding, and experimental protocols were approved by the UAH IACUC committee.
The Barnes maze setup
The Barnes maze was originally developed for rats, but works equally well for mice with a few minor modifications to the protocol (Barnes 1979; Bach et al 1995; Harrison et al 2006; Attar et al 2013; O'Leary and Brown 2013). The Barnes maze was used to assess the visual/spatial learning and long-term memory in the tyrosinemia type I mice and the mice treated with NTBC. The Barnes’ maze arena consisted of an elevated circular platform with a height of 90 cm and a 91 cm diameter (Stoelting Co., Wood Dale, IL). Around the arena’s circumference, there were 20 equally spaced holes with a 5 cm diameter. One of the 20 holes contained a dark sunken compartment that served as the escape box. In order to create a mildly aversive environment, nine 150 W spotlights were aimed at the maze in such a way to serve as motivation for the mice to navigate the maze and locate the escape box. The arena of the maze was surrounded with plastic medical screen dividers so that the mice were not distracted by the experimenters’ actions and/or unintended variable distal cues. Four proximal visual cues (a square, circle, triangle, and a cross) were attached to the clinical dividers in equal spacing so that the mice were able to orient their direction on the maze with visual cues. The mice were also able to orient themselves using spatial cues such as overhead lights, camera, cables, and ceiling tiles. The Barne’s maze protocol consisted of acclimatization, training, retention of memory, and a response to a new target hole location which we called the probe test. For details see Supplementary materials.
Data acquisition and analysis
Data were filmed, recorded, and analyzed using Ethovision XT 9.0 Software (Noldus Information Technology Inc., Leesburg, VA). Mouse movement was tracked using the geometric animal center or three point tracking using the nose, center, and tail base. Mouse movement was detected using either static or dynamic subtraction with a data sampling of 30 times per second. Data was exported as a spreadsheet and plotted using Prism 6.0 software (GraphPad Software Inc., La Jolla, CA).
Statistical analysis
All data are presented in the results as mean ± SEM unless otherwise indicated. The Barnes maze data collected from the three groups of mice, WT-water, WT-NTBC, and Fahmut-NTBC were compared using one-way ANOVA, with P < 0.05 considered significant. Intergroup analyses was performed using Tukey's multiple comparisons test, again with P < 0.05 considered significant. The graphs of primary measurements (Fig. 2a-d) are shown as mean ± SEM for visual clarity, all other graphs are presented as mean ± SD. For the urine homovanillic acid levels, data are presented as mean ± SD and were compared using a two-tailed Student’s t test. All data was plotted and statistics analyzed using Prism 6.0 software.

Mice with tyrosinemia type I take longer to learn the target hole location in the Barnes maze. Primary measured endpoints for mouse behavior were primary latency, primary distance, primary velocity and primary errors, and are shown for the three groups of test mice. In all graphs WT-water mice are represented by a solid square (■), WT-NTBC mice are represented by a solid circle (●), and the tyrosinemia type I mice (Fahmut-NTBC) are shown by a solid triangle (▲). Data are presented as mean ± SEM. A. Primary latency, the time to first find the target hole was similar for all three groups of mice on day 1. By day 2, both groups of WT mice, WT-water, and WT-NTBC, showed significant improvements in primary latency (a), primary distance (b), and primary errors (c) (P < 0.05). After two days rest all three groups of mice showed similar skill in recollection of the location of the target hole, with no difference between primary latency (a), primary distance (b), and primary errors (c). There is no difference between mouse velocity on any given day, or between any group of mice on any day (d)
Results
NTBC treatment increases the amount of homovanillic acid in the urine
NTBC inhibits the second enzyme 4-hydroxyphenylpyruvate dioxygenase (HPPD, EC 1.13.11.27) in the catabolism of tyrosine resulting in hypertyrosinemia. Tyrosine also serves as the substrate for dopamine synthesis (Fig. 1). We hypothesized that NTBC would increase the levels of the neurotransmitter/hormone dopamine, and that this may modify the behavior, learning, and memory of the mice. Mice drinking water dosed with NTBC (7.5 mg/L) had four times the amount of homovanillic acid in their urine (Supplemental Fig. 1, P < 0.0001). WT mice drinking water (WT-water) had a mean urine homovanillic acid level of 52.6 ± 5.4 μg/mg creatinine (n = 6), while the combined group of mice drinking NTBC water (WT-NTBC and Fahmut-NTBC) had a homovanillic acid level of 202.9 ± 20.0 μg/mg creatinine (n = 9). Although this is proof of concept that NTBC can increase dopamine levels, the relationship of systemic dopamine and homovanillic acid concentrations to the central nervous system dopamine levels is likely irrelevant and will be discussed.
Tyrosinemia type I mice (Fahmut-NTBC) take longer than WT-NTBC mice to learn the target hole location in the Barnes maze
We used the Barnes maze as a model to study the effect of the disease condition tyrosinemia type I and its treatment NTBC, on visual/spatial learning and long term memory in mice. On day one the primary latency to find the target hole, that is, the time it takes for the mouse to locate the target hole, but not necessarily enter the escape box, was similar between all three groups of mice, F (2, 23) = 1.559, P = 0.2318 (Fig. 2a). The WT-water mice had a primary latency of 51.43 ± 12.48 s (n = 10), the WT-NTBC mice were 78.67 ± 8.25 s (n = 8), and the Fahmut-NTBC mice were 78.92 ± 16.98 s (n = 8). However, by the second day of training the WT mice drinking water and the WT mice treated with NTBC showed dramatic improvement over the tyrosinemia type I mice in time to find the target hole, with primary latency time being 30.70 ± 4.93 s for WT-water (n = 10), 32.75 ± 6.45 s for WT-NTBC (n = 8) mice, but 98.29 ± 14.22 s (n = 8) for the Fahmut-NTBC mice, F (2, 23) = 17.90, P < 0.0001 (Fig. 2a) with both WT mice groups being similar, but the Fahmut-NTBC taking about three times longer to find the target hole (Fig. 2a, P < 0.05). This general pattern continued through day three and day four, with the tyrosinemia mice taking longer to locate the hole, but improving a little every day. The mice then had a two day maze holiday and were tested again for retention memory of the target hole location, using the visual/spatial cues. On retention day all three groups of mice were similar F (2, 23) = 2.741, P = 0.0856 (Fig. 2a), with WT-water having a primary latency of 15.40 ± 4.36 s (n = 10), WT-NTBC taking 17.13 ± 4.28 s (n = 8), and Fahmut-NTBC having a latency of 61.38 ± 28.29 s (n = 8). The large variation in the tyrosinemia mouse group was due to two wayward, unsporting mice. If these mice are excluded from the analyses, the primary latency times to target hole on retention day are essentially indistinguishable between the three groups of mice.
Tyrosinemia type I mice travel longer to find target hole in the Barnes maze
The primary distance to target hole showed a similar pattern to primary latency, but on day one WT-water mice traveled a shorter distance to find the target hole than the mice drinking NTBC (P < 0.05), F (2, 23) = 5.793, P = 0.0092 (Fig. 2b). The WT-water mice traveled 265.7 ± 48.8 cm (n = 10) while the WT-NTBC mice and Fahmut-NTBC mice traveled 551.2 ± 66.5 cm (n = 8) and 474.5 ± 76.8 cm (n = 8), respectively. On day two, both the WT-water mice and WT-NTBC mice were able to find the target hole in 219.8 ± 25.9 cm (n = 10) and 239.6 ± 46.4 cm (n = 8) respectively, while the Fahmut-NTBC mice had not improved, traveling 517.6 ± 49.8 cm (n = 8), F (2, 23) = 16.67, P < 0.0001. This learning trend continued, and after the 2 day rest, retention distances were similar between all three groups of mice, F (2, 23) = 1.877, P = 0.1757. The WT-water mice traveled 139.8 ± 29.8 cm (n = 10), while the WT-NTBC and Fahmut-NTBC mice traveled 158.3 ± 45.66 cm (n = 8) and 324.4 ± 124.5 cm (n = 8), respectively (Fig. 2b). There was no difference in primary velocity in a single group of mice during the experimental protocol or between the three groups of mice on any given day F (14, 110) = 1.406, P = 0.1624 (Fig. 2d). Using day one as an example, F (2, 22) = 0.7700, P = 0.4751 the primary velocity of WT-water mice was 6.3 ± 0.6 cm/s (n = 10) and the primary velocities of WT-NTBC and Fahmut-NTBC were 7.0 ± 0.3 cm/s (n = 8) and 7.1 ± 0.5 cm/s (n = 8) respectively (Fig. 2d).
Tyrosinemia type I mice make more errors in finding target hole in the Barnes maze
On the first day of training the mean number of errors made while finding the target hole was 3.7 ± 0.8 (n = 10) for WT-water and 8.8 ± 1.1 (n = 8) and 5.1 ± 1.0 (n = 8) for WT-NTBC and Fahmut-NTBC mice, respectively (Fig. 2c). However, by day two the tyrosinemia mice were still making 5.9 ± 0.7 errors, more than twice the errors of the WT-water and WT-NTBC mice, F (2, 23) = 12.45, P = 0.0002, which were making 2.1 ± 0.4 and 2.4 ± 0.6 errors per trial, respectively (Fig. 2c). The general trend was toward less errors in future consecutive days. On retention day, the number of errors in finding the target hole was similar between all three groups of mice, F (2, 23) = 0.7472, P = 0.4849. The number of errors made by the WT-water mice was 1.3 ± 0.4 (n = 10) and the WT-NTBC and the Fahmut-NTBC mice made 2.1 ± 0.7 (n = 8) and 2.6 ± 1.3 (n = 8) errors, respectively (Fig. 2c).
Tyrosinemia type I mice are less likely to adopt efficient search strategies
For the four training days and the retention trial, the search strategies for the three groups of mice were similar, with the mice quickly adopting the visual/spatial or the more efficient serial search strategy to find the target hole. Only the Fahmut mice did not increase their use of the efficient visual/spatial or serial search strategies over the four training days (see Supplementary materials and Supplemental Fig. 2).
The Probe experiment
The day after the retention test we performed a probe test, where we moved the location of the target hole by about 90°, either five or six holes to the left, to study how the mouse responds when it returns to the learned target location and no longer finds the escape box. We measured the primary latency to the original target hole, primary latency to the probe hole, the strategy used to find the original target hole, and then the strategy used to find the relocated probe hole once the mouse has realized that the escape box hole is no longer in its expected location. For the original target hole (Fig. 3a), there was no difference in the primary latency between all three groups of mice, F (2, 15) = 0.6332, P = 0.5445, with the WT-water mice taking 12.9 ± 5.3 s (n = 6), the WT-NTBC mice taking 11.6 ± 7.9 s (n = 4) and the Fahmut-NTBC mice 22.7 ± 8.6 s (n = 8) to find the target hole. Interestingly, four mice from each of the WT groups went straight to the vicinity of the probe hole and bypassed the target hole. These mice were excluded from the target hole analysis as they all located and immediately entered the probe hole. This probe hole was in a new and unlearned location, so we can only assume that the mice were adopting a serial search strategy and decided to start at this location and happened to stumble upon the new location of the probe hole. Alternatively, this experiment could be confounded by an as yet unrecognized variable or more likely by experimenter error.

Tyrosinemia type I mice take four times longer to find the probe hole. A. All three groups of mice found the original target hole location equally well. The WT-water mice found it in 12.9 ± 5.3 s (n = 6), the WT-NTBC mice in 11.6 ± 7.9 s (n = 4) and the Fahmut-NTBC mice in 22.7 ± 8.6 s (n = 8), with four mice in each of the WT groups finding the probe hole first, so they were excluded from this data. B. The mean time to the find the 90° relocated probe hole was 46.5 ± 14.5 s (n = 10) for WT-water and 29.9 ± 8.0 s (n = 8) for WT-NTBC, but about four times longer for the Fahmut-NTBC mice who took 120.6 ± 21.1 s (n = 8) compared to the WT-NTBC mice (P = 0.0013)
We simplified the search strategy analysis to just three classifications: visual/spatial, if the mouse travels directly to the target/probe hole, serial, if the mouse checked two or more sequential holes before finding the target/probe hole and random, if the mouse crossed the center three or more times before it finds the target/probe hole. In locating the target hole, 100 % of the WT mice used visual/spatial strategy, 75 % of the WT-water mice used visual/spatial while 25 % of the mice used serial strategy. For the Fahmut-NTBC mice 75 %, used visual/spatial strategy while 25 % of the mice used random strategy (Fig. 4a).

Tyrosinemia type I mice revert to random searching to find the moved probe hole and are less likely to enter the escape box. A. We simplified the strategy analysis to just three classifications: visual/spatial (white), serial (gray), and random (black). All of the WT-water mice (100 %) used visual/spatial searching to find the target hole, while 75 % of the NTBC mice groups used visual/spatial strategy, with the remainder of the WT-NTBC mice using serial strategy and 25 % of the Fahmut-NTBC mice using a random search strategy. When searching for the 90° shifted probe hole (P), all mice in the WT groups (WT-water and WT-NTBC) used the serial strategy, while 33 % of the mice in the Fahmut-NTBC group reverted to random searching. B. On the left, we show a representative mouse track (red line) of a WT-water mouse using a visual/spatial search strategy to locate the target hole (T), then quickly adopting a serial search strategy to find the probe hole (P) and escape box. In the right panel, we show a representative mouse track (red line) of a Fahmut-NTBC mouse that spent an extended amount of time at the previously learned target hole, before adopting a random search strategy to locate the shifted probe hole (P). C. Mice with tyrosinemia type I are less willing to enter the escape box. Mouse entries into escape box are plotted as percentage of total mice per group, per day (n = 6, four training days, one retention day and one probe day). The WT-water mice entered the escape box 75.6 ± 5.4 % (n = 10) of the time and WT-NTBC entered 81.9 ± 7.5 % (n = 8) of trials but Fahmut-NTBC mice only entered 29.9 ± 3.9 % (n = 8) of trials (P < 0.0001). D. On the left, we show a representative mouse track (red line) of a WT-water mouse using a visual/spatial search strategy to locate the target hole (T), then after a brief erroneous sojourn to the right, it adopts a serial search strategy to find the probe hole (P) and immediately enters the escape box. In the right panel, we show a representative mouse track (red line) of a Fahmut-NTBC mouse that spent an extended amount of time at the previously learned target hole visibly distressed that the escape box was not there. The tyrosinemia type I mouse then randomly searched the maze and eventually found the shifted probe hole (P), but did not enter the escape box, and even carried on exploring different holes after it located it
For the primary latency to find the new probe hole location both WT mice groups (WT-water and WT-NTBC) were similar and were four times faster than the Fahmut-NTBC mice, F (2, 23) = 9.198, P = 0.0012 (Fig. 3b). The mean time to find the probe hole was 46.5 ± 14.5 s for the WT-water mice (n = 10), it was 29.9 ± 8.0 s (n = 8) for the WT-NTBC mice and 120.6 ± 21.1 s (n = 8) for the Fahmut-NTBC mice (Fig. 3b). For the probe hole we did not assign visual/spatial strategy, as the mice had not learned the location of this hole so could not possibly use a visual/spatial strategy to locate it. All of the mice (100 %) in the WT-water group and WT-NTBC group used a serial strategy (Fig. 4a and b, left panel) to find the location of the new probe hole. While 62.5 % of the tyrosinemia type I disease Fahmut-NTBC mice used the serial strategy, the other 37.5 % used a random strategy (Fig. 4a and b, right panel).
All eight tyrosinemia type I Fahmut-NTBC mice first traveled to the original location of the target hole, and after spending an extended period of time confused as to why the escape box was no longer in that location, proceeded to search and locate the new probe hole and escape box position (Fig. 4b, right panel). More than a third of the Fahmut-NTBC mice were visibly confused and regressed to adopting a random search strategy again (Fig. 4a and b, right panel). We conclude that mice with tyrosinemia type I are not cognitively flexible and do not adapt well to change.
One unusual behavior seen in the tyrosinemia type I mice was their unwillingness to enter the target hole once they had found it, F (2, 15) = 24.09, P < 0.0001 (Fig. 4c). Calculating the percentage of hole entries for each of the six experimental days (the four training days, retention day, and probe day), the WT-water mice entered the hole 75.6 ± 5.4 % (n = 6) of the time, the WT-NTBC mice entered 81.9 ± 7.5 % (n = 6), while the Fahmut-NTBC mice entered only 29.8 ± 3.9 % (n = 6) of the time (Fig. 4c). This behavior was even more exaggerated if the probe day is analyzed separately. On probe day, 90 % of WT-water mice (9 out of 10) entered the hole, 100 % of WT-NTBC mice (8 out of 8) but only 12.5 % Fahmut-NTBC mice (1 out of 8) entered the probe hole. WT-water or WT-NTBC mice entered the escape box quickly upon finding it (Fig. 4d, left panel). While Fahmut-NTBC mice spend an extended period of time at the original target hole (Fig. 4d, right panel) before regressing into a random search strategy and eventually finding the probe hole, but these Fahmut-NTBC mice generally did not enter the escape box (Fig. 4d, right panel).
In summary, most parameters measuring visual/spatial learning and memory as assessed by the Barnes maze, were initially similar between the three groups of mice, WT-water, WT-NTBC, and the tyrosinemia type I diseased mice (Fahmut-NTBC). However, by the second day of training, the two groups of WT mice (WT-water and WT-NTBC) had improved in primary latency, primary distance, and were making fewer errors in finding the target hole than the Fahmut-NTBC. After four days of training, and a two day rest period, all three groups of mice were able to remember the location of the target hole and showed similar skills and proficiencies in finding it. The Fahmut-NTBC mice were unable to adapt to change in the probe test (shifted escape box experiment) and regressed back to a random hole searching pattern. Once the Fahmut-NTBC mice found the new location of the escape box, they spent an extended amount of time dancing around the hole and did not enter, whereas the WT-water mice or the WT-NTBC mice enter the escape box very quickly.
Discussion
One of the most pressing questions in the tyrosinemia type I field at the moment is the cause of the cognitive decline, schooling, and behavioral problems seen in children (Masurel-Paulet et al 2008; Bendadi et al 2014). The mechanism behind the cognitive decline and behavioral issues is currently unknown, and could be caused by the long term treatment with NTBC, or it could be a complication of the disease. NTBC certainly increases tyrosine levels in the plasma (Lindstedt et al 1992) and CSF (Thimm et al 2011), and tyrosine and dopamine levels in the mouse brain (Harding et al 2014).
Our findings from the Barnes maze study indicate that the Fahmut-NTBC tyrosinemia mice can learn and adopt a visual/spatial searching strategy and have no deficits in long-term memory. However, it takes them longer to learn, they make more errors, and they do not respond well to a situation of change where the target hole and escape box are moved. The Fahmut-NTBC tyrosinemia type I mice also do not enter the escape hole upon finding it. This could be due to decreased interest or motivation, less exploratory behavior, less fear (willing to remain in open), more anxiety, or more fear of the unknown (what is inside the escape box). In our future experiments we will attempt to reveal the other altered behavioral characteristics of the tyrosinemia type I condition or NTBC treatment, using additional maze and behavioral tests such as open field, elevated zero maze, novel object recognition, the Y-maze, and the Crawley three-chambered sociability test.
NTBC inhibits the enzyme 4-hydroxyphenylpyruvate dioxygenase (HPPD), inhibiting the metabolism of tyrosine at the second step, resulting in hypertyrosinemia. In looking for a cause for the cognitive, learning and behavioral changes seen in children (Bendadi et al 2014), we first addressed the importance of dopamine. We measured about a fourfold increase in homovanillic acid in the urine of both groups of mice treated with NTBC, over the mice drinking water. This is proof of concept that NTBC does increase systemic dopamine levels. Although it has been reported that increased homovanillic acid in the urine can indicate increased dopaminergic neuron activity in the central system (Pickar et al 1988), upon a closer analysis and comprehension of our data, we now believe that it is more likely that the levels of dopamine and homovanillic acid in the plasma and urine do not reflect central dopaminergic neuron activity at all, and more likely represent peripheral dopamine synthesis and breakdown (Kopin et al 1988; Lambert et al 1993).
NTBC has been shown to increase mouse brain dopamine levels by 30 %, albeit in a PKU mouse model and with a lower NTBC dose (Harding et al 2014). Therefore, it follows that NTBC will probably increase brain dopamine levels by at least 30 % in this tyrosinemia type I mouse study. This would suggest that the behavioral effects observed here are due to the absence of expression or decrease in functional activity of the FAH enzyme in the tyrosinemia type I mouse and not NTBC treatment.
After a single oral dose of radioactive NTBC in the rat, the drug concentration peaks in most tissues in about 2 hr, the drug distribution in the brain was the lowest of all tissues measured, being about 20 times less than the liver (Lock et al 1996). NTBC binds very tightly to HPPD with an IC50 of 40 nM (Ellis et al 1995). The reason for lower concentrations of NTBC found in the brain could be that NTBC is less permeable across the blood-brain barrier or that there is less HPPD expression in the brain, resulting in less NTBC bound. To explain our data we favor the theory that NTBC is poorly transported into the brain, resulting in insufficient inhibition of HPPD (Fig. 5). The target enzyme of NTBC, HPPD, is highly expressed in the brain, specifically in neurons of the cortex, cerebellum, and hippocampus (Neve et al 2003). FAH is also expressed in the brain, specifically in the white matter (Labelle et al 1993). However, there are no published data on the measured activity of either HPPD or FAH from brain. FAH was found to be highly expressed in oligodendrocytes (Labelle et al 1993), the myelin-forming glial cells of the central nervous system which maintain long-term axonal integrity (Nave 2010). If FAH is functionally present in these oligodendrocytes to metabolize tyrosine, then in its absence in tyrosinemia type I, tyrosine metabolism will proceed through to the metabolite fumarylacetoacetate and no further. Fumarylacetoacetate levels would build up and then be converted into succinylacetone, which is a neurotoxin and a potent inhibitor of δ-aminolevulinic acid dehydratase, the rate limiting enzyme in the heme synthesis pathway (Sassa and Kappas 1983). Inhibition of this enzyme results in an increase in CNS concentrations of δ-ALA which has been shown to inhibit the formation of peripheral myelin (Felitsyn et al 2008). This δ-ALA induced myelination disorder would manifest itself as a peripheral neuropathy and conduction disorder in the peripheral nervous system (Mitchell et al 1990, Gibbs et al 1993), while in the central nervous system, this hypo- or dysmyelination could manifest as a cognitive, behavioral, information processing or learning disability (Fields 2008). There are several mechanisms by which oligodendrocyte dysfunction and dysmyelination could contribute to cognitive impairment and behavioral differences, including reduced conduction velocity and conduction blocks, loss of sub millisecond synchronization of action potentials, altered axonal sprouting, and axon degeneration. δ-ALA may also cause damage to serotonergic neurons, which are extremely sensitive to oxidative damage (Nave 2010). The buildup of δ-ALA from the inhibition of heme biosynthesis can also be problematic as it can mimic GABA and bind to GABA receptors (Müller and Snyder 1977; Brennan and Cantrill 1979), resulting in a decrease of GABA receptor density in the CNS (Adhikari et al 2006).

A schematic hypothesis to explain the behavioral changes observed in tyrosinemia type I. Here we show the possible mechanisms by which tyrosinemia or NTBC could alter brain biochemistry and physiology. NTBC inhibits the catabolism of tyrosine, causing hypertyrosinemia. This high level of tyrosine will compete for transport into the brain with other large neutral amino acids (LNAA) on the SLC7 family of solute transporters. This will result in decreased uptake of other amino acids such as PHE, and TRP the precursor of serotonin. This could increase dopamine concentrations in the brain and also potentially decrease serotonin synthesis. We propose that NTBC has limited permeability across the blood-brain barrier, providing insufficient inhibition of HPPD. Hence, NTBC is less efficient at the treatment of tyrosinemia type I of the CNS. The resulting tyrosine catabolism will proceed to fumarylacetoacetate, and be blocked from proceeding further by the absence of FAH, and diverted to conversion into succinylacetone, a neurotoxin. Succinylacetone is also a potent inhibitor of δ-ALA dehydratase, an enzyme involved in the heme biosynthesis pathway. The levels of δ-ALA, another potent neurotoxin will increase, damaging oligodendrocytes and neurons, decreasing myelin synthesis and causing dys/hypo-myelination, resulting in altered cognition, learning and behavior
Conclusion
First and foremost, these data strongly suggest that the differences we see in mouse behavior in the Barnes maze are due to tyrosinemia type I and not due to treatment with NTBC. However, with our current experiments we cannot determine if NTBC treatment contributes to the cognitive and behavioral effects observed in tyrosinemia type I, but increased tyrosine levels per se have no effect on visual/spatial learning and long term memory in the wild-type mouse. We hypothesize that NTBC exhibits decreased transport into the brain and does not effectively treat tyrosinemia type I of the central nervous system (Fig. 5). This results in insufficient inhibition of HPPD and production of neurotoxic metabolites that result in a decrease in neuronal myelination. The subsequent altered white matter architecture causes the slower learning and cognitive and behavioral changes seen in tyrosinemia type I. To test our hypothesis, our future work will focus on methods to quantify and study myelin and white matter in the tyrosinemia type I mouse brain using imaging and biochemical methods. Our ultimate goal is to correlate white matter levels to cognitive changes seen in tyrosinemia type I and propose pharmaceutical interventions to correct it.
Abbreviations
- TT1:
-
Tyrosinemia type I
- FAH:
-
Fumarylacetoacetate hydrolase
- NTBC:
-
2-(2-nitro-4-trifluoromethylbenzoyl) cyclohexane-1-3-dione
- Tyr:
-
L-tyrosine
- Phe:
-
L-phenylalanine
- Trp:
-
L-tryptophan
- LNAA:
-
Large neutral amino acid
- HPPD:
-
4-hydroxyphenylpyruvate dioxygenase
- ALAD:
-
δ-aminolevulinate dehydratase
References
Adhikari A, Penatti CA, Resende RR et al (2006) 5-Aminolevulinate and 4, 5-dioxovalerate ions decrease GABA(A) receptor density in neuronal cells, synaptosomes and rat brain. Brain Res 1093(1):95–104
Aponte JL, Sega GA, Hauser LJ et al (2001) Point mutations in the murine fumarylacetoacetate hydrolase gene: Animal models for the human genetic disorder hereditary tyrosinemia type 1. Proc Natl Acad Sci U S A 98(2):641–645
Attar A, Liu T, Chan WT et al (2013) A shortened Barnes maze protocol reveals memory deficits at 4-months of age in the triple-transgenic mouse model of Alzheimer's disease. PLoS One 8(11):e80355
Bach ME, Hawkins RD, Osman M et al (1995) Impairment of spatial but not contextual memory in CaMKII mutant mice with a selective loss of hippocampal LTP in the range of the theta frequency. Cell 81(6):905–915
Barnby E (2014) Tyrosinemia type 1: an overview of nursing care. Pediatr Nurs 40(2):61–68, 90
Barnes CA (1979) Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol 93(1):74–104
Bendadi F, de Koning TJ, Visser G et al (2014) Impaired cognitive functioning in patients with tyrosinemia type I receiving nitisinone. J Pediatr 164(2):398–401
Brennan MJ, Cantrill RC (1979) Delta-aminolaevulinic acid is a potent agonist for GABA autoreceptors. Nature 280(5722):514–515
De Laet C, Munoz VT, Jaeken J et al (2011) Neuropsychological outcome of NTBC-treated patients with tyrosinaemia type 1. Dev Med Child Neurol 53(10):962–964
Deacon RM (2006) Assessing nest building in mice. Nat Protoc 1:1117–1119
Ellis MK, Whitfield AC, Gowans LA et al (1995) Inhibition of 4-hydroxyphenylpyruvate dioxygenase by 2-(2-nitro-4-trifluoromethylbenzoyl)-cyclohexane-1,3-dione and 2-(2-chloro-4-methanesulfonylbenzoyl)-cyclohexane-1,3-dione. Toxicol Appl Pharmacol 133(1):12–19
Felitsyn N, McLeod C, Shroads AL et al (2008) The heme precursor delta-aminolevulinate blocks peripheral myelin formation. J Neurochem 106(5):2068–2079
Fields RD (2008) White matter in learning, cognition and psychiatric disorders. Trends Neurosci 31(7):361–370
Gibbs TC, Payan J, Brett AM et al (1993) Peripheral neuropathy as the presenting feature of tyrosinaemia type I and effectively treated with an inhibitor of 4-hydroxyphenylpyruvate dioxygenase. J Neurol Neurosurg Psychiatry 56(10):1129–1132
Halvorsen S, Pande H, Loken AC et al (1966) Tyrosinosis. A study of 6 cases. Arch Dis Child 41(217):238–249
Harding CO, Winn SR, Gibson KM et al (2014) Pharmacologic inhibition of L-tyrosine degradation ameliorates cerebral dopamine deficiency in murine phenylketonuria (PKU). J Inherit Metab Dis 37(5):735–743
Harrison FE, Reiserer RS, Tomarken AJ et al (2006) Spatial and nonspatial escape strategies in the Barnes maze. Learn Mem 13(6):809–819
Jorquera R, Tanguay RM (2001) Fumarylacetoacetate, the metabolite accumulating in hereditary tyrosinemia, activates the ERK pathway and induces mitotic abnormalities and genomic instability. Hum Mol Genet 10(17):1741–1752
Kobayashi M, Nakamura T, Akai K (1980) A neuropathological investigation of a case of tyrosinosis. Acta Pathol Jpn 30(2):285–292
Kopin IJ, White JH, Bankiewicz K (1988) A new approach to biochemical evaluation of brain dopamine metabolism. Cell Mol Neurobiol 8(2):171–179
Labelle Y, Puymirat J, Tanguay RM (1993) Localization of cells in the rat brain expressing fumarylacetoacetate hydrolase, the deficient enzyme in hereditary tyrosinemia type 1. Biochim Biophys Acta 1180(3):250–256
Lambert GW, Eisenhofer G, Jennings GL et al (1993) Regional homovanillic acid production in humans. Life Sci 53(1):63–75
Lindblad B, Lindstedt S, Steen G (1977) On the enzymic defects in hereditary tyrosinemia. Proc Natl Acad Sci U S A 74(10):4641–4645
Lindstedt S, Holme E, Lock EA et al (1992) Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet 340(8823):813–817
Lock EA, Gaskin P, Ellis MK et al (1996) Tissue distribution of 2-(2-nitro-4-trifluoromethylbenzoyl)cyclohexane-1-3-dione (NTBC): effect on enzymes involved in tyrosine catabolism and relevance to ocular toxicity in the rat. Toxicol Appl Pharmacol 141(2):439–447
Maiorana A, Malamisura M, Emma F et al (2014) Early effect of NTBC on renal tubular dysfunction in hereditary tyrosinemia type 1. Mol Genet Metab 113(3):188–193
Masurel-Paulet A, Poggi-Bach J, Rolland MO et al (2008) NTBC treatment in tyrosinaemia type I: long-term outcome in French patients. J Inherit Metab Dis 31(1):81–87
Mitchell G, Larochelle J, Lambert M et al (1990) Neurologic crises in hereditary tyrosinemia. N Engl J Med 322(7):432–437
Müller WE, Snyder SH (1977) delta-Aminolevulinic acid: influences on synaptic GABA receptor binding may explain CNS symptoms of porphyria. Ann Neurol 2(4):340–342
Nave KA (2010) Myelination and support of axonal integrity by glia. Nature 468(7321):244–252
Neve S, Aarenstrup L, Tornehave D et al (2003) Tissue distribution, intracellular localization and proteolytic processing of rat 4-hydroxyphenylpyruvate dioxygenase. Cell Biol Int 27(8):611–624
Noble-Jamieson G, Jamieson N, Clayton P et al (1994) Neurological crisis in hereditary tyrosinaemia and complete reversal after liver transplantation. Arch Dis Child 70(6):544–545
O'Leary TP, Brown RE (2013) Optimization of apparatus design and behavioral measures for the assessment of visuo-spatial learning and memory of mice on the Barnes maze. Learn Mem 20(2):85–96
Partington MW, Haust MD (1967) A patient with tyrosinemia and hypermethioninemia. Can Med Assoc J 97(18):1059–1067
Pickar D, Breier A, Kelsoe J (1988) Plasma homovanillic acid as an index of central dopaminergic activity: studies in schizophrenic patients. Ann N Y Acad Sci 537:339–346
Pohorecka M, Biernacka M, Jakubowska-Winecka A et al (2012) Behavioral and intellectual functioning in patients with tyrosinemia type I. Pediatr Endocrinol Diabetes Metab 18(3):96–100
Santra S, Preece MA, Hulton SA et al (2008) Renal tubular function in children with tyrosinaemia type I treated with nitisinone. J Inherit Metab Dis 31(3):399–402
Sassa S, Kappas A (1983) Hereditary tyrosinemia and the heme biosynthetic pathway. Profound inhibition of delta-aminolevulinic acid dehydratase activity by succinylacetone. J Clin Invest 71(3):625–634
Thimm E, Herebian D, Assmann B et al (2011) Increase of CSF tyrosine and impaired serotonin turnover in tyrosinemia type I. Mol Genet Metab 102(2):122–125
Thimm E, Richter-Werkle R, Kamp G et al (2012) Neurocognitive outcome in patients with hypertyrosinemia type I after long-term treatment with NTBC. J Inherit Metab Dis 35(2):263–268
Van Spronsen FJ, Thomasse Y, Smit GP et al (1994) Hereditary tyrosinemia type I: a new clinical classification with difference in prognosis on dietary treatment. Hepatology 20(5):1187–1191
Wyss PA, Boynton S, Chu J et al (1993) Tissue distribution of succinylacetone in the rat in vivo: a possible basis for neurotoxicity in hereditary infantile tyrosinemia. Biochim Biophys Acta 1182(3):323–328
Acknowledgments
The authors are extremely grateful to Melanie Klarer, Wolfgang Langhans and Urs Meyer (Physiology and Behavior Laboratory, ETH Zurich, Switzerland) for teaching us the art of mouse behavioral experiments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Megan Hillgartner, Sarah Coker, Ashton Koenig, Marissa Moore, Elizabeth Barnby and Gordon MacGregor declare that they have no conflict of interest.
Animal rights
All institutional and national guidelines for the care and use of laboratory animals were followed. The care, housing, breeding, and maze experiments of animals were approved by the UAH IACUC committee.
Funding
This study was funded internally by UAH.
Additional information
Communicated by: Nenad Blau
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 494 kb)
Rights and permissions
About this article

Cite this article
Hillgartner, M.A., Coker, S.B., Koenig, A.E. et al. Tyrosinemia type I and not treatment with NTBC causes slower learning and altered behavior in mice. J Inherit Metab Dis 39, 673–682 (2016). https://doi.org/10.1007/s10545-016-9949-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-016-9949-6