
Abstract
The mitochondrial pyruvate oxidation route is a tightly regulated process, which is essential for aerobic cellular energy production. Disruption of this pathway may lead to severe neurometabolic disorders with onset in early childhood. A frequent finding in these patients is acute and chronic lactic acidemia, which is caused by increased conversion of pyruvate via the enzyme lactate dehydrogenase. Under stable clinical conditions, this process may remain well compensated and does not require specific therapy. However, especially in situations with altered energy demands, such as febrile infections or longer periods of fasting, children with mitochondrial disorders have a high risk of metabolic decompensation with exacerbation of hyperlactatemia and severe metabolic acidosis. Unfortunately, no controlled studies regarding therapy of this critical condition are available and clinical outcome is often unfavorable. Therefore, the aim of this review was to formulate expert-based suggestions for treatment of these patients, including dietary recommendations, buffering strategies and specific drug therapy. However, it is important to keep in mind that a specific therapy for the underlying metabolic cause in children with mitochondrial diseases is usually not available and symptomatic therapy especially of severe lactic acidosis has its ethical limitations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mitochondria play a central role in the production of ATP via the process of oxidative phosphorylation (OXPHOS). Basically, this biochemical system consists of the respiratory chain enzymes (mitochondrial complex I–IV), which are coupled to ATP synthase (complex V). Disruption of this pathway may lead to severe neurodegenerative disorders. The overall incidence of mitochondrial diseases is estimated at 1–5 in 10,000 live births, making this one of the commonest inherited errors of metabolism (Thorburn 2004; Schaefer et al 2008). The umbrella term “mitochondriopathy” comprises a large number of heterogeneous diseases with diverse clinical phenotypes. This aspect is further illustrated by the fact that to date, disease-causing mutations have been identified in more than 230 different genes (Koopman et al 2012; Rotig 2014). The resulting biochemical defects may affect any OXPHOS complex (isolated or as a combined deficiency). Moreover, coenzyme Q10 metabolism and pyruvate oxidation including pyruvate dehydrogenase complex (PDHC) may be impaired. In addition, secondary mitochondrial disorders may arise from an indirect impairment of OXPHOS function (for example alterations in cardiolipin metabolism as in Barth syndrome [OMIM:302060], disturbance of mitochondrial protein import as in Mohr-Tranebjaerg syndrome [OMIM:304700], inhibition of the respiratory chain in organic acidurias like propionic acidemia [OMIM:606054] or impaired uptake of cofactors of the OXPHOS system). In this review the term mitochondrial diseases is used for disorders affecting aerobic energy metabolism with a main focus on OXPHOS defects.
In general, although no good correlation exists between the biochemical defect measured in tissue samples or cultured skin fibroblasts and clinical disease expression, severe biochemical OXPHOS defects mostly present already in the neonatal period or in young infants, leading to rapid deterioration and death in early childhood (Baertling et al 2014). Clinical phenotypes include fatal neonatal lactic acidosis, classical Leigh syndrome (OMIM:256000), Leigh-like syndrome, and Alpers-Huttenlocher syndrome (OMIM:203700).
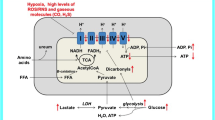
From a biochemical perspective, dysfunction of OXPHOS enzymes causes a relative increase in the production of ATP via anaerobic glycolysis, leading to intracellular accumulation of pyruvate and NADH. Under aerobic conditions, pyruvate is converted into acetyl-CoA and further oxidized via the Krebs cycle. The first irreversible reaction of glucose oxidation is catalyzed by PDHC (Mizock 1989). Therefore the activity of PDHC determines whether pyruvate is oxidized to CO2 and H2O or converted either to lactate via lactate dehydrogenase or to alanine via alanine transaminase (or alanine aminotransferase; ALAT) (Felig 1973). The activity of the PDHC is modulated by reversible phosphorylation. PDHC is inactivated by phosphorylation catalyzed by a PDH kinase (in humans there exist four distinct isoforms of this enzyme: PDK1-4), the latter of which is stimulated by ATP, NADH and acetyl-CoA. Two specific PDHC phosphatases dephosphorylate the complex and reactivate it. PDHC is further controlled by end-product inhibition (Patel and Korotchkina 2006). In the case of disturbances of the respiratory chain, NADH may accumulate in the cells (Enns et al 2000) leading to an inhibition of the PDH followed by an activation of lactate dehydrogenase, resulting in a regeneration of NAD+ and subsequent elevation of lactate (an overview of lactate metabolism is depicted in Fig. 1).

Lactate metabolism and basic concepts of small molecule interventions. Pyruvate is the end-product of glycolysis and degradation of small amino acids like alanine. In the presence of oxygen, it is transported into mitochondria via a pyruvate transporter (PT), where it is converted to acetyl-CoA catalyzed by the pyruvate dehydrogenase complex (PDHC) and finally oxidized to CO2. PDH may be inactivated through phosphorylation by pyruvate dehydrogenase kinase (PDK). PDK can be inactivated by the drug dichloroacetate (DCA). On the other hand, where there is hypoxia or impaired function of the respiratory chain, pyruvate is reduced to lactate via lactate dehydrogenase (LDH). Thus, NAD+ required for glycolysis is regenerated. Accumulated lactate is released from the cell, which may cause acidosis. The buffer solution sodium bicarbonate (Na+ + HCO3−) acts via the reaction of bicarbonate with protons. The resulting CO2 is exhaled, thereby reducing acidosis. The amine group of the buffer solution Tris(hydroxymethyl)aminomethane (THAM) takes up one proton and the protonated form of THAM is eliminated via the kidneys
Accordingly, hyperlactatemia is a common feature among children with mitochondrial disorders (Robinson 2006). Especially in situations with altered energy requirements, such as febrile infections or longer periods of fasting, patients with mitochondrial disorders have a high risk of a severe aggravation of hyperlactatemia with metabolic decompensation. Unfortunately, acute metabolic crisis is often difficult to control in these children and may have a rapidly fatal course (Koene et al 2012; Leary et al 2013). This is mainly based on the fact that treatment of lactic acidosis in mitochondrial disorders is only symptomatic and causal treatment options are very limited (Pfeffer et al 2012).
Pathophysiology of lactic acidosis
Of note, lactate per se is not toxic and there is even evidence for a neuroprotective effect (Berthet et al 2009; Wyss et al 2011). However, excessive lactate levels and severe/prolonged metabolic acidosis have finally deleterious effects on many cellular functions:
-
1)
Cardiovascular system: Increased proton concentrations in blood lead to decreased cardiac contractility, which is caused by reduced myofilament calcium sensitivity and lowered influx of calcium into the cells (Orchard and Kentish 1990). Furthermore, arterial vasodilation with simultaneous constriction of venous vessels occurs during acidosis (Mitchell et al 1972; Kellum et al 2004a, b). At the same time, acidosis leads to stimulation of the sympathetic nervous system. The functional consequences are pH dependent. At pH values above 7.2, cardiac contractility increases due to enhanced release of catecholamines. However, at pH values below 7.2, opposite effects are observed (Mitchell et al 1972). Generally, tissue response to catecholamines is decreased during acidosis (Huang et al 1995). In addition, a closure of gap junctions slows the impulse conduction of the heart and leads to a high incidence of arrhythmias (Orchard and Cingolani 1994).
-
2)
Respiratory system: By stimulation of peripheral and central chemoreceptors, breathing rate increases due to the elevated concentration of protons in order to compensate for metabolic acidosis (O’Regan and Majcherczyk 1982). The high energy consumption of this process may lead to respiratory decompensation after prolonged acidosis.
-
3)
Central nervous system (CNS): To correct intracellular acidosis, different cerebral membrane transporters are activated, leading to an intracellular accumulation of Na+ and Cl−. This results in an increased influx of water into the cells. As a consequence, cerebral edema may develop (Staub et al 1996).
-
4)
Immune system: An additional systemic effect of acidosis is a reduced immune response with increased susceptibility to serious infections caused by an elevated release of interleukins from macrophages and impaired leukocytes function (Lardner 2001; Kellum et al 2004a, b).
-
5)
Endocrine system: The response of the cells is pH dependent. Insulin resistance increases at pH <7.2 and normalizes above (Walker et al 1963). Impaired insulin sensitivity of the cells is probably caused by a pH-dependent binding of insulin to its receptor (Cuthbert and Alberti 1978).
-
6)
General effects: Multiple organs are limited in their function during prolonged acidosis, which leads to decreased tissue oxygenation, impaired ATP production and increased apoptosis (Halperin et al 1994).
Management of lactic acidosis in children with mitochondrial disorders
There are no controlled studies on treatment of lactic acidosis in children with mitochondrial diseases. Recommendations in the literature are primarily based on retrospective analyzes in children with lactic acidosis in the context of shock, sepsis or unknown underlying conditions (Hatherill et al 2003; Beveridge and Wilkinson 2006; Aschner and Poland 2008; Berg et al 2010; Parker and Parshuram 2013). While lactate elevations associated with shock or sepsis are mainly due to tissue hypoperfusion, and treatment of the underlying causes is most important to manage this condition, hyperlactatemia due to primary mitochondrial dysfunctions is caused by an impaired cellular energy metabolism with usually no causal treatment available. Accordingly, intensive care management of these patients constitutes a major challenge. Against this background, the aim of this review was to formulate expert-based suggestions for treatment of lactic acidosis in children with mitochondrial disorders. However, based on the lack of well-designed controlled studies, the level of evidence is unfortunately low. Therefore, our recommendations only represent a consensus between our clinical centers and specific details may be handled differently. Moreover, despite the treatment options described in the following sections, it is essential to keep in mind that therapy specific for the underlying metabolic cause is usually not available. Acute metabolic crisis may cause irreversible brain damage that severely affects quality of life. Therefore, decision-making regarding intensive care treatment might differ from patient to patient and should be done in close interaction with the family. If available, involvement of a specialized palliative care team should be considered.
Fluid management during metabolic crisis
Initially, normocaloric feeding and adequate fluid supplementation should be administered orally or via gastric tube. If this is not possible due to the poor clinical condition of the patient during metabolic crisis, early placement of a central venous line for buffering and parenteral nutrition is required. Catabolism should be avoided. However, infusion therapy should be done with caution since high glucose supplementation/glucose bolus application might worsen lactic acidosis (Prietsch et al 2002). As a starting point, we suggest starting infusion at a moderate glucose rate of 5–6 mg/kg/min (of note, the infusion rate might be optimized depending on age and clinical condition) (Sheridan et al 1998; Prietsch et al 2002). Serum glucose and lactate levels should be monitored closely after start of infusion therapy. After starting infusion therapy, further steps depend on close clinical re-assessment of the patient. If tolerated well, glucose rate might be further increased up to 7–8 mg/kg/min. Importantly, a parallel increase in glucose and lactate levels may indicate disturbed glucose utilization and the infusion rate should be lowered accordingly. In our experience, serum glucose levels should not exceed values between 100 and 120 mg/dl (5.6–6.7 mmol/L). The application of additional insulin to lower glucose levels and to increase cellular glucose uptake should only be used with caution because it might worsen the intracellular glucose utilization problem and aggravate acidosis. Moreover, laboratory monitoring of the critical balance between glucose and lactate levels becomes unreliable under these conditions and further therapeutic decisions may be hindered. Generally, if glucose infusion is not tolerated well, increased administration of fat (2–3 g/kg/day) should be tried to achieve adequate calorie supplementation. Of note, during the acute crisis, fat intake above 3 g/kg/day should be avoided because evidence from the literature suggests that an increased NADH/NAD+ ratio generated by reduced flux through the respiratory chain leads to a secondary inhibition of β-oxidation (Infante and Huszagh 2000). Importantly, in patients (especially neonates) where the differential diagnosis of a fatty acid oxidation disorders is not yet excluded, administration of fat should be avoided.
Specific treatment options
Unfortunately, causal treatment for children with mitochondrial diseases is usually not available. Nevertheless, specific interventions should be considered in certain conditions. In patients where the etiology of the lactic acidosis is unclear, several (not exclusively mitochondrial) metabolic diseases/transporter defects should also kept in mind. For most of the supplementations listed below no well-designed clinical trials have been performed and the results of the often anecdotic reports summarized below should be read in this context (Pfeffer et al 2013).
1) Thiamine supplementation might be effective in thiamine responsive PDHC deficiency or thiamine pyrophosphokinase deficiency (dosage: 15–30 mg/kg/day; (Mayr et al 2011)). Moreover, thiamine (and biotin) administration may be life saving in unclear disease conditions with possible differential diagnoses of thiamine deficiency (Manzanares and Hardy 2011) or thiamine metabolism disorders (biotin-thiamine-responsive basal ganglia disease; (Distelmaier et al 2013)). 2) Riboflavin supplementation was shown to improve the clinical status of patients with mitochondrial complex I deficiency caused by ACAD9 mutations (dosage: 10 mg/kg/day up to max. 300 mg/day) (Gerards et al 2011). To date the mechanism of these therapeutic effects is not sufficiently clear. It might act as cofactor and also has electron donor and acceptor properties and may thus interfere with the respiratory chain (Tarnopolsky and Raha 2005). 3) Coenzyme Q10 application should be considered in suspected coenzyme Q10 deficiency (OMIM:607426) disorders (dosage: 5–20 mg/kg/day up to max. 1 g/day) (Rotig et al 2000). To increase coenzyme Q10 oral bioavailability, application of a self-emulsifiable composition is recommended (Sato et al 2013). 4) Carnitine supplementation should be provided in cases of secondary carnitine deficiency (dosage: 50–100 mg/kg/day). 5) Ketogenic diet is well established as therapy for patients with PDHC deficiency and has neuroprotective, antiepileptic and mitochondriotropic effect also for other mitochondrial diseases (Wexler et al 1997; Klepper et al 2002). It is contraindicated in patients with known fatty acid oxidation disorders and pyruvate carboxylase deficiency. 6) Phenylbutyrate therapy might be a future option to treat lactic acidosis especially in patients with PDHC deficiency (Ferriero et al 2013). However, human studies are lacking so far. 7) Some patients with pyruvate carboxylase deficiency may profit from administration of biotin or from an anaplerotic diet containing 130 cal/kg/day with 30 % of glucose, 30 % of long-chain fatty acids, 5 % of protein, and 35 % of triheptanoin (Mochel et al 2005; Roe and Mochel 2006). Furthermore different anaplerotic substrates are tried for therapy of pyruvate carboxylase deficiency including high doses of citrate (7.5 mmol/kg/day) (Ahmad et al 1999). 8) Biotinidase deficiency can be successfully treated by administering biotin (dosage: 10–20 mg/day) (Bousounis et al 1993; Wolf 2012). For an overview see also Table 1. Details regarding mitochondrial medications and supplements are also given in the review of Parikh et al (2009).
Buffer therapy during metabolic acidosis
There is conflicting literature on the effects of infusing a buffer solution during metabolic acidosis (Kecskes and Davies 2002; Andrade et al 2007; Lim 2007; Kraut and Madias 2012). This debate is a consequence of the lack of controlled studies regarding benefits, side effects and outcome of this therapy, especially in the context of rare metabolic diseases. Nevertheless, based on the idea that severe acidosis may cause cellular tissue damage and the fact that respiratory compensation via hyperventilation is an energy consuming process, buffer therapy is frequently used during severe lactic acidosis.
Recommendations regarding timing of initiation of therapy differ in the literature and range from pH values <7.2 down to pH < 7.0 (Andrade et al 2007; Lim 2007; Sabatini and Kurtzman 2009; Kraut and Madias 2012). However, many patients are able to maintain normal pH values via excessive hyperventilation. This creates a critical condition, which might decompensate later on. Therefore, it is essential that pH values, pCO2 levels and base deficit are monitored in parallel. Importantly, rapid correction of blood pH may worsen cerebral acidosis since abrupt suppression of hyperventilation stimuli may cause increased blood pCO2, which crosses the blood brain barrier faster than HCO3 −. As a consequence, brain oedema may develop (Herrera and Kazemi 1980; Staub et al 1996). Therefore, buffering should be started rather early at a slow infusion rate. The initial hours of buffer therapy should be monitored closely with regular blood gas analyses. Of note, in situations of severe metabolic acidosis and decreased blood pressure, capillary blood sampling becomes unreliable and arterial blood gas analyses should be performed.
Sodium bicarbonate buffering
Sodium bicarbonate buffering is the most commonly used therapy for the management of acidosis due to different disorders. After injection, sodium bicarbonate dissociates to Na+ and HCO3 −. Of these two, hydrogen carbonate takes up protons to form CO2 and water. Acidosis can be subsequently reduced by exhalation of CO2 (for basic mechanism see also Fig. 1).
Of note, sodium bicarbonate buffering has several side effects, including hypernatremia, volume overload, exacerbation of intracellular acidosis, hyperosmolality and potential development of cerebral edema, hypokalemia, hypocalcaemia, and reduced cardiac contractility (Glaser et al 2001).
To minimize negative effects of sodium bicarbonate, there are several recommendations to its application. 1) If possible, isoosmotic solutions should be used, whereby hyperosmolality may be prevented (Lim 2007). However, volume overload should be avoided. 2) To minimize the increase in the intracellular acidosis, a slow application rate is recommended (see below), although there are no studies in humans available (Kecskes and Davies 2002).
To calculate the quantity of required bicarbonate volume, the distribution of bicarbonate is equated to the extracellular body fluid. In young children, this corresponds to 50–70 % of body weight. Often, the administration of 30 % is sufficient to stabilize the metabolic situation and to minimize potential side effects. This results in the following two formulas (Debray et al 2007):
or
The value of 0.3 is derived from the 30 % described above. Importantly, in neonates a value of 0.4 and in preterm infants a value of 0.5 should be inserted in the calculations. When using an undiluted 8.4 % solution for infusion (1 ml = 1 mmol sodium bicarbonate) it should be administrated through a central venous system due to its high osmolarity. Depending on the severity of acidosis and clinical condition, half of the calculated sodium bicarbonate amount can be infused over 1–2 h. The remaining amount should be infused at a slower rate. Importantly, blood gases should be monitored closely, especially during the initial buffering phase (for an overview see Table 2).
THAM (Tris-hydroxymethyl-amino-methane) buffering
THAM is a Na+-free buffer solution that can be used in conditions of respiratory or metabolic acidosis (Nahas et al 1998). Importantly, THAM does not generate increased CO2 levels, which is ideal in situations where CO2 elimination is impaired (Nahas et al 1998). During lactic acidosis, THAM reacts according to the following chemical equation: R-NH2 + H+ + Lactate− = > R-NH3 + + Lactate−. The protonated R-NH3 + is eliminated via the kidneys (for the basic mechanism see also Fig. 1).
Despite some advantages, THAM is used less frequently than bicarbonate for buffer therapy during metabolic acidosis because several potential side effects including, hyperkalaemia, hypoglycaemia, hepatotoxicity, apnoea/hypopnoea, and skin necrosis after extravasation may occur (Roberton 1970; Nahas et al 1998; Holmdahl et al 2000; Gehlbach and Schmidt 2004).
In children with mitochondrial disease and severe lactic acidosis, THAM buffering might be an alternative where the affected individual is hypernatraemic, which limits the use of bicarbonate. The THAM dosage can be calculated according to the following equation (Kraut and Madias 2012): THAM (amount in ml of a 0.3 mol/L solution) = body weight (kg) x base deficit (HCO3 − [target value] – HCO3 − [initial value]) × 1.1. It is recommended that the calculated THAM dose is infused at a slow rate (at least 1 h). The maximum daily dose is 15 mmol/kg for an adult patient. However, in newborns THAM dosage should not exceed 5–7 mmol/kg body weight because of the reduced kidney function (for an overview see Table 2).
Sodium citrate buffering
Sodium citrate buffering is mainly used for long-term treatment of patients with chronic metabolic acidosis. However, it might also be considered in the acute situation depending on the clinical condition of the patient and the wishes of the family (e.g. intravenous buffering has several side effects and may require a central venous catheter, etc.). After oral application, sodium citrate leads to a decrease in blood proton levels and a parallel increase in HCO3 − concentrations (for the defined underlying mechanism see (Kowalchuk et al 1989)). Compared with oral sodium bicarbonate application, sodium citrate does not induce gastrointestinal gas production and is usually better tolerated (McNaughton 1990). Based on our experience, therapy can be started with a dosage between 0.5 and 1.0 mmol/kg/day (divided into three doses). Further dosages should be adjusted according to blood gas analyses (for an overview see Table 2).
Dichloroacetate therapy
Dichloroacetate stimulates the enzyme pyruvate dehydrogenase kinase, and in doing so, the enzyme pyruvate dehydrogenase remains in its unphosphorylated catalytically active state (Stacpoole 1989). The resulting pyruvate is mainly converted via the Krebs cycle and lactate production decreases (for the basic mechanism see also Fig. 1). Dichloroacetate crosses the blood–brain barrier effectively and can be used in cases of chronic as well as acute lactic acidosis (Berendzen et al 2006; Kaufmann et al 2006; Stacpoole et al 2008a, b). For long-term administration in children, a dose of 25 mg/kg/d has been recommended (Abdelmalak et al 2013). A single report described the application of intravenous dichloroacetate during acute lactic acidosis in a preterm newborn with septicaemia using a dose of 50 mg/kg twice daily (Arnon et al 2001) (for an overview see Table 2). In general, dichloroacetate therapy is tolerated well in most cases and may lead to short-term improvement. However, long-term use may induce peripheral neuropathy, and there have been no studies showing clear benefits of long term DCA usage on the overall disease prognosis (Kurlemann et al 1995; Kaufmann et al 2006). A promising future option could be phenylbuytrate, which has a comparable mode of action but potentially less side effects (Ferriero et al 2013). However, clinical studies are required to evaluate this therapy.
Renal replacement therapy
Hemofiltration or hemodialysis can potentially be used for treatment of lactic acidosis. However, these are invasive procedures with the risk of side effects (see below). Moreover, they may not be feasible in unstable patients. Therefore, the use of these approaches should be critically assessed and might only be applied to exceptional cases.
In general, several studies dealing with the use of renal replacement therapy in critical situations of inborn errors of metabolism demonstrated the successful use of hemofiltration or hemodialysis to remove toxic substrates (McBryde et al 2006; Tsai et al 2014a, b). Possible advantages of using a renal replacement therapy over buffering with alkalizing substrates include the prevention of hyperosmolality and volume overload. Furthermore, the effect of alkalizing on ionized calcium concentration can be reduced by providing calcium. Renal replacement therapy leads to a quick removal of lactate but a rebound can occur. In addition, renal replacement therapy might be the treatment of choice in conditions with lactic acidosis in combination with hyperammonemia, for example in patients with mitochondrial complex V deficiency due to TMEM70 mutations (Honzik et al 2010).
Experienced personnel are required to perform renal replacement therapy, as it is technically challenging. Reliable vascular access is needed to allow adequate blood flow. The placement of catheters can be complicated by vascular stenosis, thrombosis, air emboli, or hemorrhage. A smaller vascular access in combination with lower flow rates of the blood in children increase the risk of clotting (Basu et al 2011; Bridges et al 2012). Systemic anticoagulation can result in a higher risk of developing hemorrhage. Furthermore, it might happen that the required rapid volume shifts are not well tolerated by small or critically ill children with hemodynamic instability (Tsai et al 2014a, b). Additionally, there is a risk of disequilibrium syndrome caused by an osmotic gradient between plasma and the brain because of a fast removal of urea leading to cerebral edema accompanied with potential death (Kennedy et al 1962; Arieff 1994). Thus, although renal replacement therapy has some advantages over buffering, it should only be performed in regard of the risks and by an experienced team.
Therapeutic hypothermia
Therapeutic hypothermia (TH) has been shown to be neuroprotective in newborns suffering neonatal encephalopathy after perinatal asphyxia (Jacobs et al 2013). However, the role of TH in mitochondrial disease remains unknown. Rango et al (2014) suggested that brain temperature might be reduced in patients with mitochondrial disease due to malfunctioning OXPHOS (Rango et al 2014). They hypothesized that this could be one of the reasons for impaired neurological outcome in these patients. Whether children would benefit from TH during a phase of cellular stress and severe lactic acidosis remains to be analysed in preclinical and clinical studies, and no clear recommendation can be given at this stage.
Conclusions
Metabolic crisis and severe lactic acidosis in children with mitochondrial diseases are a challenging situation. Adequate caloric supply is essential for these patients. Although discussed still somewhat controversial, buffer therapy is a mainstay of lactic acidosis management. For this purpose, different buffer solutions are available including sodium bicarbonate, THAM, and sodium citrate. Buffering should be started rather early at a slow infusion rate. In addition, dichloroacetate may be used to lower lactate levels. Of note, in certain disease conditions, the application of thiamine (alone or in combination with biotin) might improve the clinical condition (e.g. thiamine deficiency, thiamine metabolism disorders, thiamine responsive pyruvate dehydrogenase deficiency). In cases of uncontrollable lactic acidosis (especially in combination with hyperammonemia), hemofiltration therapy might be an option. Importantly, despite these symptomatic treatment options, prognosis is often unfavorable, which should be discussed openly with the relatives.
References
Abdelmalak M, Lew A, Ramezani R et al (2013) Long-term safety of dichloroacetate in congenital lactic acidosis. Mol Genet Metab 109(2):139–143
Ahmad A, Kahler SG, Kishnani PS et al (1999) Treatment of pyruvate carboxylase deficiency with high doses of citrate and aspartate. Am J Med Genet 87(4):331–338
Andrade OV, Ihara FO, Troster EJ (2007) Metabolic acidosis in childhood: why, when and how to treat. J Pediatr (Rio J) 83(2 Suppl):S11–S21
Arieff AI (1994) Dialysis disequilibrium syndrome: current concepts on pathogenesis and prevention. Kidney Int 45(3):629–635
Arnon S, Litmanovits I, Regev R, Elpeleg O, Dolfin T (2001) Dichloroacetate treatment for severe refractory metabolic acidosis during neonatal sepsis. Pediatr Infect Dis J 20(2):218–219
Aschner JL, Poland RL (2008) Sodium bicarbonate: basically useless therapy. Pediatrics 122(4):831–835
Baertling F, Rodenburg RJ, Schaper J et al (2014) A guide to diagnosis and treatment of Leigh syndrome. J Neurol Neurosurg Psychiatry 85(3):257–265
Basu RK, Wheeler DS, Goldstein S, Doughty L (2011) Acute renal replacement therapy in pediatrics. Int J Nephrol 2011:785392
Berendzen K, Theriaque DW, Shuster J, Stacpoole PW (2006) Therapeutic potential of dichloroacetate for pyruvate dehydrogenase complex deficiency. Mitochondrion 6(3):126–135
Berg CS, Barnette AR, Myers BJ, Shimony MK, Barton AW, Inder TE (2010) Sodium bicarbonate administration and outcome in preterm infants. J Pediatr 157(4):684–687
Berthet C, Lei H, Thevenet J, Gruetter R, Magistretti PJ, Hirt L (2009) Neuroprotective role of lactate after cerebral ischemia. J Cereb Blood Flow Metab 29(11):1780–1789
Beveridge CJ, Wilkinson AR (2006) Sodium bicarbonate infusion during resuscitation of infants at birth. Cochrane Database Syst Rev 1:CD004864
Bousounis DP, Camfield PR, Wolf B (1993) Reversal of brain atrophy with biotin treatment in biotinidase deficiency. Neuropediatrics 24(4):214–217
Bridges BC, Askenazi DJ, Smith J, Goldstein SL (2012) Pediatric renal replacement therapy in the intensive care unit. Blood Purif 34(2):138–148
Cuthbert C, Alberti KG (1978) Acidemia and insulin resistance in the diabetic ketoacidotic rat. Metabolism 27(12 Suppl 2):1903–1916
Debray FG, Lambert M, Chevalier I et al (2007) Long-term outcome and clinical spectrum of 73 pediatric patients with mitochondrial diseases. Pediatrics 119(4):722–733
Distelmaier F, Huppke P, Pieperhoff P, et al (2013) Biotin-responsive basal ganglia disease: a treatable differential diagnosis of leigh syndrome. JIMD reports
Enns GM, Bennett MJ, Hoppel CL et al (2000) Mitochondrial respiratory chain complex I deficiency with clinical and biochemical features of long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. J Pediatr 136(2):251–254
Felig P (1973) The glucose-alanine cycle. Metabolism 22(2):179–207
Ferriero R, Manco G, Lamantea E et al (2013) Phenylbutyrate therapy for pyruvate dehydrogenase complex deficiency and lactic acidosis. Sci Transl Med 5(175):175ra131
Gehlbach BK, Schmidt GA (2004) Bench-to-bedside review: treating acid-base abnormalities in the intensive care unit - the role of buffers. Crit Care 8(4):259–265
Gerards M, van den Bosch BJ, Danhauser K et al (2011) Riboflavin-responsive oxidative phosphorylation complex I deficiency caused by defective ACAD9: new function for an old gene. Brain 134(Pt 1):210–219
Glaser N, Barnett P, McCaslin I et al (2001) Risk factors for cerebral edema in children with diabetic ketoacidosis. The pediatric emergency medicine collaborative research committee of the American academy of pediatrics. N Engl J Med 344(4):264–269
Halperin ML, Cheema-Dhadli S, Halperin FA, Kamel KS (1994) Rationale for the use of sodium bicarbonate in a patient with lactic acidosis due to a poor cardiac output. Nephron 66(3):258–261
Hatherill M, Waggie Z, Purves L, Reynolds L, Argent A (2003) Mortality and the nature of metabolic acidosis in children with shock. Intensive Care Med 29(2):286–291
Herrera L, Kazemi H (1980) CSF bicarbonate regulation in metabolic acidosis: role of HCO3- formation in CNS. J Appl Physiol Respir Environ Exerc Physiol 49(5):778–783
Holmdahl MH, Wiklund L, Wetterberg T et al (2000) The place of THAM in the management of acidemia in clinical practice. Acta Anaesthesiol Scand 44(5):524–527
Honzik T, Tesarova M, Mayr JA et al (2010) Mitochondrial encephalocardio-myopathy with early neonatal onset due to TMEM70 mutation. Arch Dis Child 95(4):296–301
Huang YG, Wong KC, Yip WH, McJames SW, Pace NL (1995) Cardiovascular responses to graded doses of three catecholamines during lactic and hydrochloric acidosis in dogs. Br J Anaesth 74(5):583–590
Infante JP, Huszagh VA (2000) Secondary carnitine deficiency and impaired docosahexaenoic (22:6n-3) acid synthesis: a common denominator in the pathophysiology of diseases of oxidative phosphorylation and beta-oxidation. FEBS Lett 468(1):1–5
Jacobs SE, Berg M, Hunt R, Tarnow-Mordi WO, Inder TE, Davis PG (2013) Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev 1:CD003311
Kaufmann P, Engelstad K, Wei Y et al (2006) Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology 66(3):324–330
Kecskes ZB, Davies MW (2002) Rapid correction of early metabolic acidaemia in comparison with placebo, no intervention or slow correction in LBW infants. Cochrane Database Syst Rev 1:CD002976
Kellum JA, Song M, Li J (2004a) Science review: extracellular acidosis and the immune response: clinical and physiologic implications. Crit Care 8(5):331–336
Kellum JA, Song M, Venkataraman R (2004b) Effects of hyperchloremic acidosis on arterial pressure and circulating inflammatory molecules in experimental sepsis. Chest 125(1):243–248
Kennedy AC, Linton AL, Eaton JC (1962) Urea levels in cerebrospinal fluid after haemodialysis. Lancet 1(7226):410–411
Klepper J, Leiendecker B, Bredahl R et al (2002) Introduction of a ketogenic diet in young infants. J Inherit Metab Dis 25(6):449–460
Koene S, Rodenburg RJ, van der Knaap MS et al (2012) Natural disease course and genotype-phenotype correlations in Complex I deficiency caused by nuclear gene defects: what we learned from 130 cases. J Inherit Metab Dis 35(5):737–747
Koopman WJ, Willems PH, Smeitink JA (2012) Monogenic mitochondrial disorders. N Engl J Med 366(12):1132–1141
Kowalchuk JM, Maltais SA, Yamaji K, Hughson RL (1989) The effect of citrate loading on exercise performance, acid-base balance and metabolism. Eur J Appl Physiol Occup Physiol 58(8):858–864
Kraut JA, Madias NE (2012) Treatment of acute metabolic acidosis: a pathophysiologic approach. Nat Rev Nephrol 8(10):589–601
Kurlemann G, Paetzke I, Moller H et al (1995) Therapy of complex I deficiency: peripheral neuropathy during dichloroacetate therapy. Eur J Pediatr 154(11):928–932
Lardner A (2001) The effects of extracellular pH on immune function. J Leukoc Biol 69(4):522–530
Leary SC, Antonicka H, Sasarman F et al (2013) Novel Mutations in SCO1 as a cause of fatal infantile encephalopathy and lactic acidosis. Hum Mutat 34(10):1366–1370
Lim S (2007) Metabolic acidosis. Acta Med Indones 39(3):145–150
Manzanares W, Hardy G (2011) Thiamine supplementation in the critically ill. Curr Opin Clin Nutr Metab Care 14(6):610–617
Mayr JA, Freisinger P, Schlachter K et al (2011) Thiamine pyrophosphokinase deficiency in encephalopathic children with defects in the pyruvate oxidation pathway. Am J Hum Genet 89(6):806–812
McBryde KD, Kershaw DB, Bunchman TE et al (2006) Renal replacement therapy in the treatment of confirmed or suspected inborn errors of metabolism. J Pediatr 148(6):770–778
McNaughton LR (1990) Sodium citrate and anaerobic performance: implications of dosage. Eur J Appl Physiol Occup Physiol 61(5–6):392–397
Mitchell JH, Wildenthal K, Johnson RL Jr (1972) The effects of acid-base disturbances on cardiovascular and pulmonary function. Kidney Int 1(5):375–389
Mizock BA (1989) Lactic acidosis. Dis Mon 35(4):233–300
Mochel F, DeLonlay P, Touati G et al (2005) Pyruvate carboxylase deficiency: clinical and biochemical response to anaplerotic diet therapy. Mol Genet Metab 84(4):305–312
Nahas GG, Sutin KM, Fermon C et al (1998) Guidelines for the treatment of acidaemia with THAM. Drugs 55(2):191–224
O’Regan RG, Majcherczyk S (1982) Role of peripheral chemoreceptors and central chemosensitivity in the regulation of respiration and circulation. J Exp Biol 100:23–40
Orchard CH, Cingolani HE (1994) Acidosis and arrhythmias in cardiac muscle. Cardiovasc Res 28(9):1312–1319
Orchard CH, Kentish JC (1990) Effects of changes of pH on the contractile function of cardiac muscle. Am J Physiol 258(6 Pt 1):C967–C981
Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R, Medicine Society TM (2009) A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol 11(6):414–30
Parker MJ, Parshuram CS (2013) Sodium bicarbonate use in shock and cardiac arrest: attitudes of pediatric acute care physicians*. Crit Care Med 41(9):2188–2195
Patel MS, Korotchkina LG (2006) Regulation of the pyruvate dehydrogenase complex. Biochem Soc Trans 34(Pt 2):217–222
Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF (2012) Treatment for mitochondrial disorders. Cochrane Database Syst Rev 4:CD004426
Pfeffer G, Horvath R, Klopstock T et al (2013) New treatments for mitochondrial disease-no time to drop our standards. Nat Rev Neurol 9(8):474–481
Prietsch V, Lindner M, Zschocke J, Nyhan WL, Hoffmann GF (2002) Emergency management of inherited metabolic diseases. J Inherit Metab Dis 25(7):531–546
Rango M, Arighi A, Bonifati C, Del Bo R, Comi G, Bresolin N (2014) The brain is hypothermic in patients with mitochondrial diseases. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 34(5):915–920
Roberton NR (1970) Apnoea after THAM administration in the newborn. Arch Dis Child 45(240):206–214
Robinson BH (2006) Lactic acidemia and mitochondrial disease. Mol Genet Metab 89(1–2):3–13
Roe CR, Mochel F (2006) Anaplerotic diet therapy in inherited metabolic disease: therapeutic potential. J Inherit Metab Dis 29(2–3):332–340
Rotig A (2014) Genetics of mitochondrial respiratory chain deficiencies. Revue neurologique
Rotig A, Appelkvist EL, Geromel V et al (2000) Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet 356(9227):391–395
Sabatini S, Kurtzman NA (2009) Bicarbonate therapy in severe metabolic acidosis. J Am Soc Nephrol 20(4):692–695
Sato Y, Mutoh H, Suzuki M, Takekuma Y, Iseki K, Sugawara M (2013) Emulsification using highly hydrophilic surfactants improves the absorption of orally administered coenzyme Q10. Biol Pharm Bull 36(12):2012–2017
Schaefer AM, McFarland R, Blakely EL et al (2008) Prevalence of mitochondrial DNA disease in adults. Ann Neurol 63(1):35–39
Sheridan RL, Yu YM, Prelack K, Young VR, Burke JF, Tompkins RG (1998) Maximal parenteral glucose oxidation in hypermetabolic young children: a stable isotope study. JPEN J Parenter Enteral Nutr 22(4):212–216
Stacpoole PW (1989) The pharmacology of dichloroacetate. Metab Clin Exp 38(11):1124–1144
Stacpoole PW, Gilbert LR, Neiberger RE et al (2008a) Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics 121(5):e1223–e1228
Stacpoole PW, Kurtz TL, Han Z, Langaee T (2008b) Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev 60(13–14):1478–1487
Staub F, Winkler A, Haberstok J et al (1996) Swelling, intracellular acidosis, and damage of glial cells. Acta Neurochir Suppl 66:56–62
Tarnopolsky MA, Raha S (2005) Mitochondrial myopathies: diagnosis, exercise intolerance, and treatment options. Med Sci Sports Exerc 37(12):2086–2093
Thorburn DR (2004) Mitochondrial disorders: prevalence, myths and advances. J Inherit Metab Dis 27(3):349–362
Tsai IJ, Hwu WL, Huang SC et al (2014) Efficacy and safety of intermittent hemodialysis in infants and young children with inborn errors of metabolism. Pediatr Nephrol 29(1):111–116
Walker BG, Phear DN, Martin FI, Baird CW (1963) Inhibition of insulin by acidosis. Lancet 2(7315):964–965
Wexler ID, Hemalatha SG, McConnell J et al (1997) Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets. Studies in patients with identical mutations. Neurology 49(6):1655–1661
Wolf B (2012) Biotinidase deficiency: “if you have to have an inherited metabolic disease, this is the one to have”. Genet Med Off J Am Coll Med Genet 14(6):565–575
Wyss MT, Jolivet R, Buck A, Magistretti PJ, Weber B (2011) In vivo evidence for lactate as a neuronal energy source. J Neurosci 31(20):7477–7485
Funding source
This project was supported by the BMBF funded German Network for Mitochondrial Disorders (mitoNET #01GM1113C).
Conflict of interest
Katharina Danhauser, Peter Freisinger, Wolfgang Sperl, Hemmen Sabir, Dirk Klee, Berit Hadzik, Ertan Mayatepek, Eva Morava and Felix Distelmaier declare that they have no conflict of interest. Jan Smeitink is founder and CEO of Khondrion, a university spin-off company of the Radboud University Medical Centre, Nijmegen, The Netherlands.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Verena Peters
Rights and permissions
About this article

Cite this article
Danhauser, K., Smeitink, J.A.M., Freisinger, P. et al. Treatment options for lactic acidosis and metabolic crisis in children with mitochondrial disease. J Inherit Metab Dis 38, 467–475 (2015). https://doi.org/10.1007/s10545-014-9796-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-014-9796-2