
Abstract
Patients with glycogen storage diseases type 1 (GSD1) suffer from life-threatening hypoglycaemia, when left untreated. Despite an intensive dietary treatment, patients develop severe complications, such as liver tumors and renal failure, with aging. Until now, the animal models available for studying the GSD1 did not survive after weaning. To gain further insights into the molecular mechanisms of the disease and to evaluate potential treatment strategies, we have recently developed novel mouse models in which the catalytic subunit of glucose-6 phosphatase (G6pc) is deleted in each glucose-producing organ specifically. For that, B6.G6pcex3lox/ex3lox mice were crossed with transgenic mice expressing a recombinase under the control of the serum albumin, the kidney androgen protein or the villin promoter, in order to obtain liver, kidney or intestine G6pc−/− mice, respectively. As opposed to total G6pc knockout mice, tissue-specific G6pc deficiency allows mice to maintain their blood glucose by inducing glucose production in the other gluconeogenic organs. Even though it is considered that glucose is produced mainly by the liver, liver G6pc−/− mice are perfectly viable and exhibit the same hepatic pathological features as GSD1 patients, including the late development of hepatocellular adenomas and carcinomas. Interestingly, renal G6pc−/− mice developed renal symptoms similar to the early human GSD1 nephropathy. This includes glycogen overload that leads to nephromegaly and morphological and functional alterations in the kidneys. Thus, our data suggest that renal G6Pase deficiency per se is sufficient to induce the renal pathology of GSD1. Therefore, these new mouse models should allow us to improve the strategies of treatment on both nutritional and pharmacological points of view.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
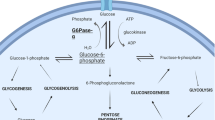
Glycogen storage disease type 1 (GSD1) is a rare metabolic disorder characterized by the absence of endogenous glucose production, leading to severe hypoglycemia following a short fast (Chou et al 2010; Froissart et al 2011). This is caused by a deficiency of glucose-6 phosphatase (G6Pase), which is an enzyme complex involving the glucose-6 phosphate translocase subunit (G6PT, encoded by SLC37A4) and the G6Pase catalytic subunit, encoded by G6PC1 (Soty et al 2012) (Fig. 1). Patients with GSD type 1a (GSD1a) represent 80 % of all GSD1 cases and have a G6PC deficiency, whereas patients with GSD type 1b (GSD1b) have a G6PT defect. While G6PT is ubiquitously expressed, G6PC is expressed only in the liver, kidneys and intestine (Rajas et al 1999, 2007; Mithieux et al 2004b). Both GSD1a and GSD1b patients show broadly similar symptoms, including severe hypoglycemia in the post-absorptive state, hyperlipidemia, hypercholesterolemia, hyperuricemia and lactic acidemia. G6Pase deficiency leads to the accumulation of glycogen and triglycerides in the liver and kidneys. This results in marked hepatomegaly, nephromegaly and hepatic steatosis. In addition, patients with GSD1b present severe infectious complications, due to neutropenia and neutrophil and monocyte functional defects.

Schematic representation of the glucose-6 phosphatase. This enzyme is composed of two subunits localized in the membrane of the endoplasmic reticulum (ER): the transport subunit (G6PT) and the catalytic subunit (G6PC). The G6PT subunit is ubiquitously expressed, whereas the G6PC is expressed only in the liver, kidneys and intestine. The mutations of G6PC are responsible for GSD type 1a and the mutations of G6PT are responsible for GSD type 1b. Images were made with Servier Medical Art illustrations
As of today, there is no cure for GSD1. In the past, many patients with GSD1 did not survive infancy and childhood. Since the 1980s, life expectancy of patients with GSD1 has been considerably improved by stringent dietary treatment (Rake et al 2002a; Heller et al 2008). Frequent meals combined with uncooked cornstarch (during the day- and/or night-time) or gastric drip-feeding allow patients with GSD1 to avoid hypoglycemia and lactic acidemia. Despite the intensive dietary treatment, hepatic, renal, and intestinal complications arise with aging (Di Rocco et al 2008; Reddy et al 2009). Thus a large proportion of patients older than 20 years show hepatic tumors and/or progressive chronic renal disease, which are the leading causes of morbidity in GSD1 patients with aging. The first hepatic tumors generally appear during adolescence and are mainly classified as hepatocellular adenomas (HCA), which can transform into hepatocellular carcinomas (HCC) (Rake et al 2002b; Franco et al 2005; Wang et al 2011; Calderaro et al 2013). Tumor resection or liver transplantation are recommended if the tumors are associated with serious compression or hemorrhage, or show signs of malignant transformation into HCC (Rake et al 2002a; Reddy et al 2009). The first symptoms of renal disease are hyperperfusion and hyperfiltration and generally appear from childhood (Martens et al 2009). Almost all adult patients show albuminuria and more than 50 % present proteinuria (Martens et al 2009). Finally, renal disease can slowly progress into renal failure that requires renal dialysis or transplantation (Rake et al 2002a).
In order to investigate the onset of long-term pathologies developed by patients with GSD1a and to evaluate potential treatment strategies, we recently developed new mouse models of GSD1a that exclusively target G6PC deletion in the liver, kidneys or intestine (Mutel et al 2011a; Penhoat et al 2011; Clar et al 2014). It is noteworthy that these tissue-specific G6PC knockout mice have normal life expectancy, whereas total G6PC knockout mouse and canine models die rapidly after weaning in the absence of intensive dietary therapy. In this review, we propose an overview of these new murine GSD1a models with tissue-specific G6pc deletions.
The canine and mouse models of total G6pc deficiency
Until now, the understanding of the biochemical bases of GSD1a and the evaluation of gene therapy approaches to correct G6pc deficiency were performed in two animal models of GSD1a. Both of these animal models are physiologically similar to humans in regard of the glucose-6 phosphate metabolism. After the isolation of the G6pc gene, Dr. Janice Chou created the G6pc knockout mouse in 1993 (Lei et al 1996). These mice present low birth weight, develop quickly severe and unremitting hypoglycemia, display quickly and gradually pronounced increases in serum cholesterol and triglyceride levels (Table 1). However, they do not typically manifest lactic acidemia. Moreover, only 60 % of mice treated with glucose injections, glucose-fortified water and food supplementation survive through weaning and have a life expectancy of about 6 months. Although all G6pc knockout mice developed hepatomegaly and steatosis (Table 1), no hepatic tumors have been reported, even in mice as old as 6 months (Salganik et al 2009). These mice exhibited nephromegaly as well (Table 1). Nevertheless, the renal pathology has been characterized only in an early stage (in 6-week-old KO mice) (Yiu et al 2008). A GSD1a canine model (Maltese breed) carrying a natural G6pc mutation was identified and used to characterize the disease as well (Kishnani et al 1997). The Maltese breed is small in size and exhibits low survival rate of newborns. A second canine model was obtained by crossbreeding a carrier Maltese with beagles (Kishnani et al 2001). Both canine models manifest all of the typical symptoms of the human disorder, including hyperlactacidemia, but they did not prove useful for studying the long-term complications of the GSD1a. Indeed, the dietary therapies used to maintain GSD1a animals viable have not yet been sufficiently refined to prevent premature death of these animals from hypoglycemia.
Rationale underlying the generation of organ-specific G6pc deficient mice
The maintenance of blood glucose levels within a narrow range (about 5.5 mmol/L) is a critical physiological function. Although the liver is the main glucose-producing organ in the post-absorptive state via glycogenolysis, renal, and intestinal glucose productions play a major role in maintaining normoglycemia during long fasting periods (Ekberg et al 1999; Gerich et al 2001; Croset et al 2001; Mithieux et al 2004a). We hypothesized that total inactivation of G6pc in only one gluconeogenic tissue would not be lethal due to the compensatory induction of glucose production by the two non-targeted tissues. To delete the G6Pase activity, we targeted the excision of G6pc exon 3, by using B6.G6pcex3lox/ex3lox mice. We crossed B6.G6pcex3lox/ex3lox mice with mice expressing a CRE recombinase under a tissue-specific promoter to target the G6pc deletion in the liver, kidney or intestine specifically. As endogenous glucose production, especially in the liver, is critical during the neonatal period because of the low content of glucose in milk (Girard et al 1992; Chatelain et al 1998), we chose to induce G6pc deletion in adult mice by using a CREERT2 inducible by tamoxifen. The CREERT2 is a recombinant CRE, fused to a mutated ligand-binding protein of the estrogen, resulting in a tamoxifen-dependent CRE. In order to induce G6pc deletion, adult mice are treated daily with 1 mg of tamoxifen for 5 consecutive days.
Liver G6pc knockout mice
Hepatic G6pc deletion was targeted by crossing B6.G6pcex3lox/ex3lox mice with B6.SACREERT2/wmice, which expressed the CREERT2 under the liver-specific serum albumin promoter. Rapidly (10 days) after tamoxifen treatment, hepatic G6Pase activity was undetectable in liver-specific G6pc knockout (L.G6pc−/−) mice (Mutel et al 2011a). As expected, L.G6pc−/− mice had normal life expectancy and showed normal blood glucose during fed state (Table 1). Interestingly, L.G6pc−/− mice did not exhibit marked hypoglycemia during long fasting periods thanks to the induction of extrahepatic glucose production (Mutel et al 2011b; Penhoat et al 2014). The livers of L.G6pc−/− mice were rapidly enlarged, due to the accumulation of glycogen and triglycerides (Table 1). In parallel, there was a rapid increase in plasma triglyceride, cholesterol, uric acid, and lactic acid levels (Table 1). However, these plasmatic parameters (except for cholesterol) improved after 6 months, as observed in total G6pc knockout mice, which survive weaning thanks to a diet therapy (Salganik et al 2009). We suggested that a satisfying blood glucose control could explain this amelioration. This is also in line with the observation that metabolic control is easier in adult patients with GSD1a, than during infancy or childhood.
Contrary to plasmatic parameters, glycogen storages were still elevated after 18 months of G6pc deletion. It is noteworthy that the accumulation of triglycerides increased with time. This could be linked to the late development of hepatocellular adenomas observed in L.G6pc−/− mice fed a standard diet. The first millimetric nodules were detected by magnetic resonance imaging after 9 months of G6pc deletion in about 20 % of L.G6pc−/− mice. Most of them developed multiple nodules of about 1 to 10 mm in diameter after 18 months of G6pc deletion. Most of these tumors were HCA, but some L.G6pc−/− livers (20 %) presented dysplasia as well (Mutel et al 2011a). In addition, about 5–10 % of L.G6pc−/− mice fed a standard diet developed HCC (unpublished data). It is important to note that the development of tumors appeared rather late, while liver steatosis tended to worsen. Moreover, our recent data show that the development of hepatic tumors in L.G6pc−/− mice is enhanced by a high fat enriched diet (to be published). To propose therapeutic strategies or to update the dietary advices, it is now important to determine the molecular mechanisms involved in tumor development and to understand how is tumorigenesis influenced by the hepatic metabolism. Thus L.G6pc−/− mice would be a unique model that could allow us to answer these questions and to study the effect of diet on the development of HCA and HCC.
Kidney G6pc knockout mice
Renal G6pc deletion was targeted by crossing B6.G6pcex3lox/ex3lox mice with B6.KapCREERT2/w mice, which expressed the CREERT2 under the kidney androgen-regulated protein (Kap) promoter (Clar et al 2014). This resulted in a partial lost (50 % of inhibition) of G6Pase activity after tamoxifen treatment of adult male mice. Immunohistological observations of K-G6pc−/− kidneys revealed a lower and heterogeneous staining of G6PC in the cortex with the presence of only a few G6PC-positive cells identified in the proximal convoluted tubules. Female B6.G6pcex3lox/ex3lox.KapCREERT2/w mice have to be treated with both testosterone and tamoxifen to induce G6pc deletion. As expected, K-G6pc−/− mice did not suffer from hypoglycemia and did not require diet treatment. This is partially due to the residual renal G6Pase activity and to a compensatory induction of hepatic G6Pase activity (Clar et al 2014). It is noteworthy that K-G6pc−/− mice showed an early stage nephropathy, i.e., microalbuminuria and a partial electrolyte imbalance, after 6 months of G6pc deletion (Table 1) (Clar et al 2014). However, they did not develop proteinuria, kidney fibrosis, and nephrolithiasis. On a molecular level, the accumulation of glucose-6 phosphate activated the de novo lipogenesis pathway. The slight accumulation of triglycerides observed in the K-G6pc−/− kidney by red Soudan staining was sufficient to activate the renin-angiotensin system, which was associated with an increased expression of the transforming growth factor β1 (TGFβ1). This led to partial epithelial-mesenchymal transition (EMT)-like changes, highlighted by a decrease of epithelial marker expression (e.g., E-cadherin, tight junction ZO-1) and an overexpression of the mesenchymal marker fibronectin. Podocyte injury characterized by a decrease in podocin, synaptopodin, and podocalyxin expression was observed in K-G6pc−/− kidneys. However, no modifications in the thickness of basement membrane or glomerulosclerosis were observed, suggesting that K-G6pc−/− kidneys showed an early stage of EMT only after 6 months of G6pc deletion (Clar et al 2014).
The characterization of renal K-G6pc−/− metabolism allowed us to highlight similarities of molecular pathways involved in the development of EMT in both GSD1 and diabetes (Mundy and Lee 2002; Rajas et al 2013). Indeed, the accumulation of lipids or lipid derivatives leads to the activation of renin- angiotensin system and subsequently of the TGFβ1 pathway. The presence of lipid deposits in the renal cortex of two GSD1 patients with proteinuria was reported in only one study (Obara et al 1993). As opposed to diabetic nephropathy, renal lipid accumulation in K-G6pc−/− mice did not seem to be mediated by the transcriptional factors SREBP1 (sterol regulatory element-binding protein), but rather by ChREBP (carbohydrate-responsive element binding protein) (Clar et al 2014). Compared to the liver, little is known about lipid metabolism and lipid deposits in the kidney. The K-G6pc−/− mouse model is therefore a unique tool that can be used to decipher the role of lipids, compared to glycogen accumulation, in the development of nephropathy.
Intestinal G6pc knockout mice
Until now, intestinal symptoms were often underestimated in GSD1a patients, when compared to GSD1b in whom they appear more serious (Visser et al 2002). However, some studies reported that GSD1a patients might also suffer from intermittent diarrhea due to entero-proctitis (Fine et al 1969; Milla et al 1978; Rake et al 2002b). An abnormal accumulation of glycogen has also been reported in the intestine of GSD1a patients (Field et al 1965).
Intestinal G6pc deletion was targeted by crossing B6.G6pcex3lox/ex3lox mice with B6.VillinCREERT2/w mice, which express the CREERT2 under the Villin promoter (Penhoat et al 2011). The specific deletion of G6pc in the intestine of I-G6pc−/− mice was persistent for more than one year. During the first year after G6pc gene deletion, I-G6pc−/− mice exhibited a growth rate similar to that of wild-type mice. Meanwhile, no diarrhea or abnormal consistency of the stools was observed in any I-G6pc−/− mice, compared to wild-type mice. Histology analysis of I-G6pc−/− proximal (duodenum) and more distal (jejunum and ileum) parts of intestine did not show glycogen or lipid accumulation. No inflammation was observed along the whole intestine after one year of deletion. However, the quality of diet can influence intestine metabolism. Hence, we cannot exclude that some adverse effects could appear with time according to the diet composition. This hypothesis will be assessed in the I-G6pc−/− mice in the near future.
For example, the ingestion of too much sugar, including uncooked starched, in patients with GSD1 could lead to bacterial proliferation. A bacterial overgrowth has been documented in a GSD1b patient by using a hydrogen breath test (Santer et al 2003). In the latter study, the authors suggested that the absence of a functional G6Pase might induce carbohydrate malabsorption by the intestine and promote bacterial overgrowth. Indeed, G6Pase has been suggested to be involved in intestinal transepithelial glucose transport. The transepithelial glucose transport in the intestine is thought to mainly involve the sodium-dependent glucose cotransporter SGLT1 at the apical membrane and the glucose transporter GLUT2 at the basal membrane. However, glucose absorption, challenged by an oral glucose test, is similar in Glut2−/− mice and wild-type mice, suggesting the existence of another intestinal glucose transport pathway (Stümpel et al 2001). The absorption of glucose measured in isolated perfused intestine and liver of Glut2−/− mice is inhibited by S4048, a specific inhibitor of G6PT. In the absence of GLUT2, a step of phosphorylation (by hexokinase) and hydrolysis (by G6Pase) seems thus needed for glucose transport through the intestine. This would be consistent with a high hexokinase activity in the intestine, tenfold greater than the reported maximal flux of glucose through glycolysis (Newsholme and Carrié 1994). Finally, the gut microbiota fermentation is tightly coupled to intestinal metabolism (De Vadder et al 2014). I-G6pc−/− mice are thus a unique model that can be used to study the precise role of intestinal glucose absorption and metabolism in the etiology of intestinal symptoms of GSD1 patients.
Lessons from the tissue-specific G6pc knockout mouse models
The usefulness of these new animal models of GSD1a is based primarily on the fact that these animals are viable. They permitted the characterization of the development of hepatic tumors and nephropathy during several months. These results suggest that the hepatic or renal G6pc deficiency per se is sufficient to induce independently the development of hepatic or renal pathology, respectively. This highlighted the fact that the development of nephropathy is independent of the liver-derived metabolic changes. This was an important finding in the context of the pathophysiological concept of the GSD1 disease as a whole, but also in terms of transplantation or gene therapy. Kidney transplantation is performed in the case of severe renal failure, but this does not correct hypoglycemia (Emmett and Narins 1978). In contrast, liver transplantation allows a correction of the glycemia and all liver-related biochemical abnormalities, but any beneficial effects on the kidney remain to be proven (Faivre et al 1999; Labrune 2002; Davis and Weinstein 2008; Reddy et al 2009). This finding suggests that clinicians should discuss the long-term benefits and possible convenience of a double kidney/liver transplantation for GSD1a patients, who have developed multiple liver HCAs or HCCs or renal failure.
Concerning gene therapy, it was already shown that the rescue of G6Pase activity in the liver by using an AAV8 recombinant vector allowed total G6pc KO mice to maintain normal blood glucose levels, but it did not prevent the development of the nephropathy (Yiu et al 2010; Luo et al 2011). As AAV1 or AAV2/9 vectors are able to transduce both the liver and the kidney, these vectors seem to be a better choice for GSD1a gene therapy (Ghosh et al 2006; Luo et al 2011). Thus, the hepatic and renal G6pc−/− mouse models will be useful to test the efficiency and the safety of gene therapy. In addition, L.G6pc−/− mice could be used to test drugs targeting the development of hepatic tumors. Interestingly, the molecular mechanisms involved in GSD1a nephropathy are very similar to those of diabetic patients (Mundy and Lee 2002; Rajas et al 2013). Therefore, K.G6pc−/− mice could be a useful tool in future studies of the pharmacological treatment of EMT and/or kidney failure developed by patients with either GSD1 or diabetes.
Finally, these mice should allow us to improve the strategies of treatment on a nutritional point of view. Indeed, preliminary results showed that diet could greatly influence the development of hepatic tumors in L.G6pc−/− mice (to be published). Moreover, until now, dietary guidelines were only based on biochemical knowledge and vary greatly between pediatricians. For example, some clinicians prohibit entirely the consumption of fruits, juice, dairy products, and sweets. Others only limit the consumption of these products, in order to restrain the intake of fructose, lactose, and sucrose. To provide scientific evidence, L.G6pc−/− and K.G6pc−/− mice will be useful to analyze the effect of diet on the development of hepatic tumors and nephropathy, respectively. Meanwhile, it is important to remind teenagers and adults to strictly follow their diet, even if they already exhibit a good metabolic control, in order to delay or avoid the development of the GSD1 complications.
References
Calderaro J, Labrune P, Morcrette G et al (2013) Molecular characterization of hepatocellular adenomas developed in patients with glycogen storage disease type I. J Hepatol 58:350–357. doi:10.1016/j.jhep.2012.09.030
Chatelain F, Pégorier JP, Minassian C et al (1998) Development and regulation of glucose-6-phosphatase gene expression in rat liver, intestine, and kidney: in vivo and in vitro studies in cultured fetal hepatocytes. Diabetes 47:882–889
Chou JY, Jun HS, Mansfield BC (2010) Glycogen storage disease type I and G6Pase-β deficiency: etiology and therapy. Nat Rev Endocrinol 6:676–688. doi:10.1038/nrendo.2010.189
Clar J, Gri B, Calderaro J et al (2014) Targeted deletion of kidney glucose-6 phosphatase leads to nephropathy. Kidney Int. doi:10.1038/ki.2014.102
Croset M, Rajas F, Zitoun C et al (2001) Rat small intestine is an insulin-sensitive gluconeogenic organ. Diabetes 50:740–746
Davis MK, Weinstein DA (2008) Liver transplantation in children with glycogen storage disease: controversies and evaluation of the risk/benefit of this procedure. Pediatr Transplant 12:137–145. doi:10.1111/j.1399-3046.2007.00803.x
De Vadder F, Kovatcheva-Datchary P, Goncalves D et al (2014) Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 156:84–96. doi:10.1016/j.cell.2013.12.016
Di Rocco M, Calevo MG, Taro’ M et al (2008) Hepatocellular adenoma and metabolic balance in patients with type Ia glycogen storage disease. Mol Genet Metab 93:398–402. doi:10.1016/j.ymgme.2007.10.134
Ekberg K, Landau BR, Wajngot A et al (1999) Contributions by kidney and liver to glucose production in the postabsorptive state and after 60 h of fasting. Diabetes 48:292–298
Emmett M, Narins RG (1978) Renal tranplantation in type 1 glycogenosis. Failure to improve glucose metabolism. JAMA J Am Med Assoc 239:1642–1644
Faivre L, Houssin D, Valayer J et al (1999) Long-term outcome of liver transplantation in patients with glycogen storage disease type Ia. J Inherit Metab Dis 22:723–732
Field JB, Epstein S, Egan T (1965) Studies in glycogen storage diseases. I. Intestinal glucose-6 phosphatase activity in patients with Von Gierke’s disease and their parents. J Clin Invest 44:1240–1247. doi:10.1172/JCI105230
Fine RN, Kogut MD, Donnell GN (1969) Intestinal absorption in type I glycogen storage disease. J Pediatr 75:632–635
Franco LM, Krishnamurthy V, Bali D et al (2005) Hepatocellular carcinoma in glycogen storage disease type Ia: a case series. J Inherit Metab Dis 28:153–162. doi:10.1007/s10545-005-7500-2
Froissart R, Piraud M, Boudjemline AM et al (2011) Glucose-6-phosphatase deficiency. Orphanet J Rare Dis 6:27. doi:10.1186/1750-1172-6-27
Gerich JE, Meyer C, Woerle HJ, Stumvoll M (2001) Renal gluconeogenesis: its importance in human glucose homeostasis. Diabetes Care 24:382–391
Ghosh A, Allamarvdasht M, Pan C-J et al (2006) Long-term correction of murine glycogen storage disease type Ia by recombinant adeno-associated virus-1-mediated gene transfer. Gene Ther 13:321–329. doi:10.1038/sj.gt.3302650
Girard J, Ferré P, Pégorier JP, Duée PH (1992) Adaptations of glucose and fatty acid metabolism during perinatal period and suckling-weaning transition. Physiol Rev 72:507–562
Heller S, Worona L, Consuelo A (2008) Nutritional therapy for glycogen storage diseases. J Pediatr Gastroenterol Nutr 47(Suppl 1):S15–S21. doi:10.1097/MPG.0b013e3181818ea5
Kim SY, Weinstein DA, Starost MF et al (2008) Necrotic foci, elevated chemokines and infiltrating neutrophils in the liver of glycogen storage disease type Ia. J Hepatol 48:479–485. doi:10.1016/j.jhep.2007.11.014
Kishnani PS, Bao Y, Wu JY et al (1997) Isolation and nucleotide sequence of canine glucose-6-phosphatase mRNA: identification of mutation in puppies with glycogen storage disease type Ia. Biochem Mol Med 61:168–177
Kishnani PS, Faulkner E, VanCamp S et al (2001) Canine model and genomic structural organization of glycogen storage disease type Ia (GSD Ia). Vet Pathol 38:83–91
Labrune P (2002) Glycogen storage disease type I: indications for liver and/or kidney transplantation. Eur J Pediatr 161(Suppl 1):S53–S55. doi:10.1007/s00431-002-1004-y
Lei KJ, Chen H, Pan CJ et al (1996) Glucose-6-phosphatase dependent substrate transport in the glycogen storage disease type-1a mouse. Nat Genet 13:203–209. doi:10.1038/ng0696-203
Luo X, Hall G, Li S et al (2011) Hepatorenal correction in murine glycogen storage disease type I with a double-stranded adeno-associated virus vector. Mol Ther J Am Soc Gene Ther 19:1961–1970. doi:10.1038/mt.2011.126
Martens DHJ, Rake JP, Navis G et al (2009) Renal function in glycogen storage disease type I, natural course, and renopreservative effects of ACE inhibition. Clin J Am Soc Nephrol CJASN 4:1741–1746. doi:10.2215/CJN.00050109
Milla PJ, Atherton DA, Leonard JV et al (1978) Disordered intestinal function in glycogen storage disease. J Inherit Metab Dis 1:155–157
Mithieux G, Bady I, Gautier A et al (2004a) Induction of control genes in intestinal gluconeogenesis is sequential during fasting and maximal in diabetes. Am J Physiol Endocrinol Metab 286:E370–E375. doi:10.1152/ajpendo.00299.2003
Mithieux G, Rajas F, Gautier-Stein A (2004b) A novel role for glucose 6-phosphatase in the small intestine in the control of glucose homeostasis. J Biol Chem 279:44231–44234. doi:10.1074/jbc.R400011200
Mundy HR, Lee PJ (2002) Glycogenosis type I and diabetes mellitus: a common mechanism for renal dysfunction? Med Hypotheses 59:110–114
Mutel E, Abdul-Wahed A, Ramamonjisoa N et al (2011a) Targeted deletion of liver glucose-6 phosphatase mimics glycogen storage disease type 1a including development of multiple adenomas. J Hepatol 54:529–537. doi:10.1016/j.jhep.2010.08.014
Mutel E, Gautier-Stein A, Abdul-Wahed A et al (2011b) Control of blood glucose in the absence of hepatic glucose production during prolonged fasting in mice: induction of renal and intestinal gluconeogenesis by glucagon. Diabetes 60:3121–3131. doi:10.2337/db11-0571
Newsholme EA, Carrié AL (1994) Quantitative aspects of glucose and glutamine metabolism by intestinal cells. Gut 35:S13–S17
Obara K, Saito T, Sato H et al (1993) Renal histology in two adult patients with type I glycogen storage disease. Clin Nephrol 39:59–64
Penhoat A, Mutel E, Amigo-Correig M et al (2011) Protein-induced satiety is abolished in the absence of intestinal gluconeogenesis. Physiol Behav 105:89–93. doi:10.1016/j.physbeh.2011.03.012
Penhoat A, Fayard L, Stefanutti A et al (2014) Intestinal gluconeogenesis is crucial to maintain a physiological fasting glycemia in the absence of hepatic glucose production in mice. Metabolism 63:104–111. doi:10.1016/j.metabol.2013.09.005
Rajas F, Bruni N, Montano S et al (1999) The glucose-6 phosphatase gene is expressed in human and rat small intestine: regulation of expression in fasted and diabetic rats. Gastroenterology 117:132–139
Rajas F, Jourdan-Pineau H, Stefanutti A et al (2007) Immunocytochemical localization of glucose 6-phosphatase and cytosolic phosphoenolpyruvate carboxykinase in gluconeogenic tissues reveals unsuspected metabolic zonation. Histochem Cell Biol 127:555–565. doi:10.1007/s00418-006-0263-5
Rajas F, Labrune P, Mithieux G (2013) Glycogen storage disease type 1 and diabetes: learning by comparing and contrasting the two disorders. Diabetes Metab. doi:10.1016/j.diabet.2013.03.002
Rake JP, Visser G, Labrune P et al (2002a) Guidelines for management of glycogen storage disease type I - European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr 161(Suppl 1):S112–S119. doi:10.1007/s00431-002-1016-7
Rake JP, Visser G, Labrune P et al (2002b) Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr 161(Suppl 1):S20–S34. doi:10.1007/s00431-002-0999-4
Reddy SK, Austin SL, Spencer-Manzon M et al (2009) Liver transplantation for glycogen storage disease type Ia. J Hepatol 51:483–490. doi:10.1016/j.jhep.2009.05.026
Salganik SV, Weinstein DA, Shupe TD et al (2009) A detailed characterization of the adult mouse model of glycogen storage disease Ia. Lab Investig J Tech Methods Pathol 89:1032–1042. doi:10.1038/labinvest.2009.64
Santer R, Hillebrand G, Steinmann B, Schaub J (2003) Intestinal glucose transport: evidence for a membrane traffic-based pathway in humans. Gastroenterology 124:34–39. doi:10.1053/gast.2003.50009
Soty M, Chilloux J, Casteras S et al (2012) New insights into the organisation and intracellular localisation of the two subunits of glucose-6-phosphatase. Biochimie 94:695–703. doi:10.1016/j.biochi.2011.09.022
Stümpel F, Burcelin R, Jungermann K, Thorens B (2001) Normal kinetics of intestinal glucose absorption in the absence of GLUT2: evidence for a transport pathway requiring glucose phosphorylation and transfer into the endoplasmic reticulum. Proc Natl Acad Sci U S A 98:11330–11335. doi:10.1073/pnas.211357698
Visser G, Rake JP, Kokke FTM et al (2002) Intestinal function in glycogen storage disease type I. J Inherit Metab Dis 25:261–267
Wang DQ, Fiske LM, Carreras CT, Weinstein DA (2011) Natural history of hepatocellular adenoma formation in glycogen storage disease type I. J Pediatr 159:442–446. doi:10.1016/j.jpeds.2011.02.031
Yiu WH, Pan C-J, Ruef RA et al (2008) Angiotensin mediates renal fibrosis in the nephropathy of glycogen storage disease type Ia. Kidney Int 73:716–723. doi:10.1038/sj.ki.5002718
Yiu WH, Lee YM, Peng W-T et al (2010) Complete normalization of hepatic G6PC deficiency in murine glycogen storage disease type Ia using gene therapy. Mol Ther J Am Soc Gene Ther 18:1076–1084. doi:10.1038/mt.2010.64
Financial support
This work was supported by research grants from the “AgenceNationale de la Recherche” (ANR-07-MRAR-011-01 and ANR11-BSV1-009-01) and the “Association Francophone des Glycogénoses”.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Georg Hoffmann
Rights and permissions
About this article

Cite this article
Rajas, F., Clar, J., Gautier-Stein, A. et al. Lessons from new mouse models of glycogen storage disease type 1a in relation to the time course and organ specificity of the disease. J Inherit Metab Dis 38, 521–527 (2015). https://doi.org/10.1007/s10545-014-9761-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-014-9761-0