
Abstract
Disorders of the glucose-6-phosphatase (G6Pase)/glucose-6-phosphate transporter (G6PT) complexes consist of three subtypes: glycogen storage disease type Ia (GSD-Ia), deficient in the liver/kidney/intestine-restricted G6Pase-α (or G6PC); GSD-Ib, deficient in a ubiquitously expressed G6PT (or SLC37A4); and G6Pase-β deficiency or severe congenital neutropenia syndrome type 4 (SCN4), deficient in the ubiquitously expressed G6Pase-β (or G6PC3). G6Pase-α and G6Pase-β are glucose-6-phosphate (G6P) hydrolases with active sites lying inside the endoplasmic reticulum (ER) lumen and as such are dependent upon the G6PT to translocate G6P from the cytoplasm into the lumen. The tissue expression profiles of the G6Pase enzymes dictate the disease's phenotype. A functional G6Pase-α/G6PT complex maintains interprandial glucose homeostasis, while a functional G6Pase-β/G6PT complex maintains neutrophil/macrophage energy homeostasis and functionality. G6Pase-β deficiency is not a glycogen storage disease but biochemically it is a GSD-I related syndrome (GSD-Irs). GSD-Ia and GSD-Ib patients manifest a common metabolic phenotype of impaired blood glucose homeostasis not shared by GSD-Irs. GSD-Ib and GSD-Irs patients manifest a common myeloid phenotype of neutropenia and neutrophil/macrophage dysfunction not shared by GSD-Ia. While a disruption of the activity of the G6Pase-α/G6PT complex readily explains why GSD-Ia and GSD-Ib patients exhibit impaired glucose homeostasis, the basis for neutropenia and myeloid dysfunction in GSD-Ib and GSD-Irs are only now starting to be understood. Animal models of all three disorders are now available and are being exploited to both delineate the disease more precisely and develop new treatment approaches, including gene therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
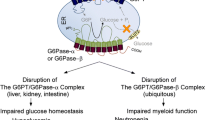
Glycogen storage disease type I (GSD-I), also known as von Gierke disease, is a group of autosomal recessive disorders caused by deficiencies in the activities of the glucose-6-phosphatase (G6Pase)/glucose-6-phosphate transporter (G6PT) complexes (Chou et al 2002, 2010: Chou and Mansfield 2014). The incidence of GSD-I is 1 in 100,000 live births. Early studies suggested there were four subtypes, GSD-Ia, GSD-Ib, GSD-Ic, and GSD-Id, but it is now well established that GSD-Ib is implicated in all reported cases of GSD-I that are not GSD-Ia (Chou et al 2010). GSD-Ia (MIM232200) is caused by a deficiency in the liver/kidney/intestine-restricted G6Pase-α (or G6PC) while GSD-Ib (MIM232220) is caused by a deficiency in the ubiquitously expressed G6PT (or SLC37A4) (Fig. 1). Prior to 2003 only a liver/kidney/intestine-restricted G6Pase activity had been identified, which was referred to as simply G6Pase. More recently, with the identification of a second ubiquitously expressed G6Pase isoform (Shieh et al 2003), the nomenclature has changed. The original G6Pase is now called G6Pase-α and the ubiquitous activity is called G6Pase-β (or G6PC3) (Shieh et al 2003). Both G6Pase-α (Lei et al 1993) and G6Pase-β (Shieh et al 2003) catalyze the hydrolysis of glucose-6-phosphate (G6P) to glucose and phosphate and both are key enzymes for intracellular glucose production (Fig. 1). Topological analyses showed that both G6Pase-α (Pan et al 1998; Ghosh et al 2002) and G6Pase-β (Shieh et al 2003; Ghosh et al 2004) span the endoplasmic reticulum (ER) membrane, with multiple domains, their active sites lying inside the ER lumen. Therefore for G6P catalysis, both depend on the ER-associated transmembrane protein G6PT to translocate G6P from the cytoplasm into the ER lumen and each must couple functionally with G6PT to form a G6Pase/G6PT complex. Since G6PT is expressed ubiquitously, the different phenotypes in the GSD-I diseases reflect the tissue expression profiles of G6Pase-α or G6Pase-β and the resulting G6Pase/G6PT complexes (Fig. 1).

GSD-Ia, GSD-Ib, and GSD-Irs manifest distinct and overlapping phenotypes. G6Pase-α, G6PT, and G6Pase-β are shown embedded within the membrane of the ER
A functional G6Pase-α/G6PT complex maintains interprandial blood glucose homeostasis while a functional G6Pase-β/G6PT complex maintains energy homeostasis and functionality in neutrophils and macrophages (Chou et al 2002, 2010; Jun et al 2012). Mutations of G6Pase-α underlie GSD-Ia which has a metabolic phenotype of impaired blood glucose homeostasis, characterized by hypoglycemia, hepatomegaly, nephromegaly, hypertriglyceridemia, hypercholesterolemia, hyperuricemia, lactic acidemia, and growth retardation (Fig. 1) (Chou et al 2002, 2010). Mutations in G6Pase-β underlie a severe congenital neutropenia syndrome type 4 (SCN4, MIM612541), characterized by neutropenia and neutrophil/macrophage dysfunction (Fig. 1) (Cheung et al 2007; Boztug et al 2009; Jun et al 2010, 2012; McDermott et al 2010; Banka and Newman 2013). The latter includes impairment in respiratory burst, chemotaxis, calcium mobilization, and phagocytosis activities. Mutations of G6PT underlie GSD-Ib, which shares the same metabolic phenotype of impaired glucose homeostasis with GSD-Ia but carries the additional complications of neutropenia and myeloid dysfunction typical of G6Pase-β deficiency (Chou et al 2002, 2010; Kim et al 2008; Jun et al 2014). While G6Pase-β deficiency does not have an immediately apparent glycogen storage phenotype typical of GSD-Ia and GSD-Ib, it is a disease of the G6Pase/G6PT complex. Whether there is a glycogen storage issue in the myeloid cells remains to be determined. We propose the name GSD-I related syndrome (GSD-Irs) for G6Pase-β deficiency to reflect the biochemical and functional relationship to GSD-I. While the molecular bases for GSD-Ia, GSD-Ib, and GSD-Irs are now well established, many aspects of the diseases are still poorly understood. This review focuses on recent developments elucidating the molecular mechanisms underlying neutropenia and myeloid dysfunction in GSD-Ib and GSD-Irs as well as on the recombinant adeno-associated virus (rAAV) vector-mediated gene therapy for the treatment of GSD-Ia. Clinical therapies for GSD-Ia and GSD-Ib have been extensively reviewed (Visser et al 2002; Koeberl et al 2009; Chou et al 2010; Shah and O'Dell 2013; Boers et al 2014) and will only be briefly summarized in this review.
GSD-Ia
GSD-Ia (G6Pase-α deficiency) is the most prevalent form of type I GSD, representing ~ 80 % of cases (Chou et al 2002, 2010). Human G6PC is a single copy gene composed of five exons, located on chromosome 17q21 (Lei et al 1993; Chou et al 2002) that is expressed primarily in the liver, kidney, and intestine. The encoded G6Pase-α enzyme is a 357 amino-acid glycoprotein anchored to the ER by nine transmembrane helices (Pan et al 1998). Based on mutational and active site labeling studies, the current paradigm for the G6Pase-α reaction mechanism is that His-176 initiates a nucleophilic attack on the phosphate of G6P to form a phosphohistidine-enzyme intermediate (Ghosh et al 2002). This transition state is stabilized byArg-83 hydrogen bonding to phosphate, and is resolved by His-119 providing a proton that liberates the glucose moiety. The active site residues, Arg-83, His-119, and His-176 are all situated inside the ER lumen inaccessible to G6P in the cytoplasm. Eighty-nine separate G6PC mutations, including 58 missense, ten nonsense, 17 insertion/deletion, three splicing (Chou et al 2002, 2010; Chou and Mansfield 2008), and one no-stop mutation (c.1074A > C/p.358Yext43) have been identified to date [http://www.hgmd.cf.ac.uk/ac/gene.php?gene=G6PC]. Of the identified mutations, 50 missense, two nonsense, and two codon deletion mutations have been confirmed as pathogenic using site-directed mutagenesis and transient expression assays (Shieh et al 2002; Chou and Mansfield 2008). Thirty-two of the missense mutations completely abolish G6Pase-α activity and the other 18 retain varying degrees of residual enzymatic activity (Shieh et al 2002; Chou and Mansfield 2008). While GSD-Ia is not predominantly restricted to any one racial or ethnic group, mutations in the G6PC gene apparently unique to Caucasian, Hispanic, Chinese/Japanese/Korean, and Jewish GSD-Ia patients have been described (Chou et al 2002, 2010) suggesting separate ethnic founder effects for some mutations. GSD-Ia is more prevalent in the Ashkenazi Jewish population where the carrier frequency for the p.R83C mutation is 1.4 % (Ekstein et al 2004). To date, no clear genotype-phenotype correlations have been demonstrated in GSD-Ia (Matern et al 2002; Chou et al 2010).
GSD-Ib
GSD-Ib (G6PT deficiency) represents ~20 % of GSD-I cases (Chou et al 2002, 2010). The human G6PT protein is encoded by the single copy SLC37A4 gene that was initially reported, based on sequence homology, as a member of the solute carrier family 37 (SLC37) (Chen et al 2008; Chou and Mansfield 2014). The SLC37A4 gene is composed of nine exons (Hiraiwa et al 1999) located on chromosome 11q23 (Annabi et al 1998) and produces two alternatively spliced transcripts, that encode the primary, ubiquitously expressed G6PT (Gerin et al 1997) and the brain/heart/muscle-specific variant G6PT (Lin et al 2000). The significance of these expression patterns is not understood as both proteins are catalytically active, span the ER membrane with ten domains (Pan et al 1999; Lin et al 2000). As predicted, G6PT has been demonstrated to be a sugar-phosphate/phosphate exchanger and functions as an antiporter — a phosphate-linked transporter that exchanges cytoplasmic G6P for inorganic phosphate stored in the lumen of the ER (Chen et al 2008). The primary in vivo function of the G6PT protein is to translocate G6P from the cytoplasm into the ER lumen, delivering it to the catalytic site of G6Pase-α or G6Pase-β for hydrolysis into glucose and phosphate (Chou et al 2002, 2010). This transport activity is dependent on the ability of G6PT to form a functional complex with a G6Pase. In the absence of a G6Pase, the G6P transport activity is minimal (Lei et al 1996; Hiraiwa et al 1999; Chen et al 2008). Consequently, G6PT is essential to maintain both interprandial blood glucose homeostasis and myeloid cell energy homeostasis, making GSD-Ib both a metabolic and immune disorder.
GSD-Ib is not restricted to any one racial or ethnic group, although prevalent mutations in the SLC37A4 gene have been described in several groups (Chou et al 2002, 2010; Chou and Mansfield 2014). Notably, not all patients diagnosed with GSD-Ib, based on their metabolic phenotype and genetic analysis, seem to develop neutropenia (Chou et al 2010). Whether this is a true lack of penetrance, which might suggest there are important modifier gene activities, or not, remains to be determined. To date, 92 separate mutations have been identified in the SLC37A4 gene of GSD-Ib and non-GSD-Ia patients (Chou et al 2010; Chou and Mansfield 2014). These include 39 missense, 11 nonsense, 22 insertion/deletion, and 19 splicing mutations. Of these, 31 missense and two codon-deletion mutations have been confirmed as pathogenic using site-directed mutagenesis and transient expression assays, resulting in the abolishment, or greatly reduced microsomal G6P uptake activity (Chou et al 2002; Chou and Mansfield 2014). As with the G6PC mutations there does not appear to be a strict genotype-phenotype relationship.
GSD-Irs
GSD-Irs (G6Pase-β deficiency) is a rare, autosomal recessive neutropenia syndrome, with a prevalence less than one in 1 million, identified in 2009 (Boztug et al 2009). The human, single-copy G6PC3 gene consists of six exons on chromosome 17q21 and encodes a highly hydrophobic, 346-amino acid G6Pase-β polypeptide (Martin et al 2002). Despite marked structural similarity with G6Pase-α, the two enzymes share only 36 % amino acid homology (Shieh et al 2003). Similar to G6Pase-α, G6Pase-β spans the ER membrane with nine transmembrane domains (Ghosh et al 2004). Sequence alignment with G6Pase-α suggests the active center of G6Pase-β is comprised of Arg-79, His-114, and His-167, which lie inside the ER lumen (Shieh et al 2003; Ghosh et al 2004). Site-directed mutagenesis and transient expression assays support this notion, as mutations in any of these proposed catalytic-site residues abolish enzyme activity (Shieh et al 2003). Active-site labeling has also established His-167 as the phosphate acceptor that forms the phosphohistidine-G6Pase-β intermediate during catalysis (Ghosh et al 2004), analogous to His-176 in G6Pase-α.
GSD-Irs presents with the same neutropenia and myeloid dysfunctions seen in GSD-Ib (Cheung et al 2007; Boztug et al 2009; Jun et al 2010; McDermott et al 2010). However, the syndrome also presents with non-haematological defects, including prominent superficial venous pattern, congenital cardiac anomaly, and genital anomalies (Boztug et al 2009; Banka and Newman 2013), not observed in GSD-Ib patients. This may point to additional roles for G6Pase-β that are not yet characterized. Twenty-nine separate mutations, including 15 missense, four nonsense, three splicing, and seven insertions and/or deletions, have been identified (Banka and Newman 2013). To date, only the p.R253H and p.G260R mutations have been characterized functionally and shown to be pathogenic (Boztug et al 2009; McDermott et al 2010). However, neither the yeast expression system used for p.R253H (Boztug et al 2009) nor the Epstein-Barr virus-transformed lymphoblastoid cell line assay used for p.G260R (McDermott et al 2010) are optimal for the assay of the low activity G6Pase-β. Functional characterization in a more sensitive, low background assay, such as the adenoviral expression system used for characterizing the active-site mutants of G6Pase-β, should give more definitive results (Shieh et al 2003).
Animal models
Several animal models of GSD-Ia exist, including a GSD-Ia knockout mouse (G6pc−/−) (Lei et al 1996), a naturally occurring dog model (Kishnani et al 2001), and two conditional G6pc-null mouse models (Peng et al 2009; Mutel et al 2011). The G6pc−/− mice manifests all of the known symptoms of human GSD-Ia, although it has a relatively mild lactic acidemia compared to human GSD-Ia patients (Lei et al 1996). The reason for the lactate difference remains to be elucidated, but the mouse still appears to be an excellent model of the human disease. The GSD-Ia dog model is based on a naturally occurring p.M121I G6PC mutation identified in the Maltese breed that was cross-bred into Beagles to overcome the size, neonatal survival, and small litter size limitations of the carrier Maltese background (Kishnani et al 2001). This Maltese-Beagle hybrid manifests all the typical symptoms of the human disorder, including a marked lactic acidosis more typical of human GSD-Ia patients.
For GSD-Ib and GSD-Irs, only transgenic knockout mouse models are currently available. The mouse model for GSD-Ib (G6pt−/−) exhibits all the metabolic and immune abnormalities of human GSD-Ib, including impaired glucose homeostasis, neutropenia and defects in neutrophil respiratory burst, chemotaxis, calcium mobilization, and phagocytotic activities (Chen et al 2003). The mouse model for GSD-Irs (G6pc3−/−) manifests both neutropenia and myeloid dysfunction of human GSD-Ib and GSD-Irs (Cheung et al 2007). Human GSD-Irs patients also exhibit non-haematological defects (reviewed in Banka and Newman 2013). Since GSD-Irs is a newly identified syndrome, whether the G6pc3−/− mice manifest the non-haematological defects seen in human GSD-Irs patients remains to be elucidated.
These animal models have been widely used to understand the biology, pathophysiology, and long-term complications of the GSD-I disorders. They have proved invaluable in delineating the molecular mechanisms underlying myeloid dysfunction in GSD-Ib and GSD-Irs, and have been exploited to develop gene and cell therapies of GSD-I. The G6pc−/− mice, G6pc−/− dogs, and G6pt−/− mice rarely survive longer than 3 months even under intensive dietary therapy regimes. Consequently the presence and natural history of many other aspects of the human diseases, including inflammatory bowel disease (IBD) or enterocolitis in GSD-Ib, are difficult to study and have not yet been investigated. However, hepatocellular adenomas (HCA) can be studied using the conditional liver-specific G6pc-null (L-G6pc−/−) mice which are viable and develop hepatomegaly, hepatic steatosis, and multiple HCA (Mutel et al 2011). To delineate the molecular mechanisms underlying other long-term complications of these disorders, a mouse with conditional knockout in the kidney or intestine may be of value.
Metabolic phenotype — GSD-Ia and GSD-Ib
GSD-Ia and GSD-Ib share a common metabolic phenotype, the hallmark of which is hypoglycemia following a short fast (Chou et al 2002, 2010). The liver, and to a lesser extent, the kidney and intestine, are the primary gluconeogenic organs involved in the regulation of blood glucose homeostasis between meals. As blood glucose levels fall between meals, an increase in cytoplasmic G6P produced in the terminal step of gluconeogenesis and glycogenolysis in the liver, kidney, and intestine is transported by G6PT into the ER where it is hydrolyzed by G6Pase-α to glucose and released into the blood (Fig. 2). The defective G6Pase-α/G6PT complex impairs this process, creates elevated levels of cytoplasmic G6P, and fails to maintain blood glucose homeostasis. The elevated cytoplasmic G6P drives glycogen accumulation, leading to hepatomegaly and nephromegaly. Hepatomegaly is further exacerbated by an accumulation of liver neutral lipids. Other major metabolic consequences of elevated G6P are hyperlipidemia, hyperuricemia, and lactic acidemia that characterize the clinical pathophysiology of GSD-Ia and GSD-Ib (Chou et al 2002, 2010). Longer-term presentations include osteoporosis, gout, pulmonary hypertension, renal disease, and HCA that may undergo malignant transformation to hepatocellular carcinoma (Chou et al 2002, 2010; Rake et al 2002; Franco et al 2005).

The primary anabolic and catabolic pathways of G6P in the gluconeogenic organs, liver, kidney, and intestine. These three organs are the primary sources for the production of intracellular glucose via hydrolysis of G6P produced in the terminal rate-limiting step of gluconeogensis and glycogenolysis by G6Pase-α. They are responsible for maintaining interprandial blood glucose homeostasis. A simplified cell is shown containing an enlargement of the ER. G6Pase-α and G6PT are shown embedded within the membrane of the ER. GLUT2, the transporter responsible for the transport of glucose in and out of the cell in liver, kidney, and intestine, is shown embedded in the plasma membrane
Myeloid phenotype — GSD-Ib and GSD-Irs
The mechanism underlying clinical neutropenia and myeloid dysfunction in patients with GSD-Ib and GSD-Irs results from a defective G6Pase-β/G6PT complex in myeloid tissues (Chou et al 2010). This complex is dysfunctional if either G6Pase-β or G6PT loses activity, therefore studies of either GSD-Ib or GSD-Irs provides information about the other. Kuijpers et al (2003) showed that neutrophils of GSD-Ib patients exhibit enhanced apoptosis, suggesting a causal relationship between apoptosis and neutropenia, but its underlying cause remained undetermined. With the discovery of G6Pase-β and its functional coupling with G6PT (Shieh et al 2003), the question arose whether endogenous glucose production by the G6Pase-β/G6PTcomplex in the ER of neutrophils was critical for neutrophil homeostasis and function. Cheung et al (2007) showed that neutrophils from G6pc3−/− mice, which are unable to hydrolyze endolumenal G6P to glucose, also exhibit enhanced ER stress and apoptosis. Likewise, neutrophils from G6pt−/−mice, which are unable to translocate G6P from the cytoplasm into the lumen of the ER, display enhanced ER stress and apoptosis (Kim et al 2008). Supporting this, neutrophils of GSD-Irs patients were shown to exhibit enhanced ER stress and apoptosis (Boztug et al 2009; Jun et al 2010; McDermott et al 2010). Taken together, these data support the hypothesis that enhanced neutrophil apoptosis underlies, at least in part, neutropenia in GSD-Ib and GSD-Irs. Supporting this further, neutrophil apoptosis in both G6pc3−/− (Jun et al 2011) and G6pt−/− (Kim et al 2008) mice was shown to be mediated by the intrinsic mitochondrial stress pathway.
In addition to neutropenia, both GSD-Ib (Kim et al 2008; Jun et al 2014) and GSD-Irs (Cheung et al 2007; Jun et al 2010; McDermott et al 2010) also exhibit neutrophil dysfunction characterized by impaired chemotaxis, calcium mobilization, respiratory burst, and phagocytic activities. IBD, indistinguishable from idiopathic Crohn disease is also a clinical presentation of both disorders (Chou et al 2010; Banka and Newman 2013). The molecular mechanisms underlying neutrophil dysfunction in GSD-Ib and GSD-Irs are now becoming understood. Neutrophils require a constant supply of glucose for both function and survival, yet are unable to produce glucose via gluconeogenesis. Therefore, the primary source of glucose is uptake from the blood. There are three primary pathways that compete for intracellular glucose/G6P in neutrophils, namely: glycolysis; the hexose monophosphate shunt (HMS); and ER cycling of G6P/glucose (Jun et al 2010) (Fig. 3). The latter pathway is mediated by the G6Pase-β/G6PT complex (Jun et al 2010). Both G6PT-deficient (Jun et al 2014) and G6Pase-β-deficient (Jun et al 2010) neutrophils exhibit impaired neutrophil glucose uptake. In parallel, levels of G6P, lactate, and ATP are markedly lower in G6PT-deficient (Jun et al 2014) and G6Pase-β-deficient (Jun et al 2010) neutrophils, compared with the controls. Moreover, the expression and activation of NADPH oxidase, a multi-component enzyme that facilitates the production of reactive oxygen species is down-regulated in neutrophils of both disorders (Jun et al 2010, 2014). Consequently, inactivation of either component of the G6Pase-β/G6PT complex disrupts neutrophil energy homeostasis, leading to impaired neutrophil respiratory burst, chemotaxis, calcium flux, and phagocytosis activities.

Pathways for G6P metabolism in neutrophils. A simplified cell is shown containing an enlargement of the ER. Glucose transported into the cytoplasm via GLUT1 is metabolized by hexokinase to G6P which can participate in three major pathways: glycolysis, HMS or ER cycling. In cycling, G6P enters the ER via G6PT where it can accumulate until it is hydrolyzed to glucose by G6Pase-β and transported back into the cytoplasm. By limiting the cytoplasmic glucose/G6P availability, cycling regulates the other two cytoplasmic pathways for G6P metabolism. Disruption of this cycling in G6PT- or G6Pase-β-deficient neutrophils results in impaired energy homeostasis and functionality. The GLUT1 transporter, responsible for the transport of glucose in and out of the cell, is shown embedded in the plasma membrane. G6PT and G6Pase-β are shown embedded in the ER membrane
GSD-Ib patients also manifest monocyte/macrophage dysfunction (Kilpatrick et al 1990; McCawley et al 1993). Using G6pc3−/− mice, Jun et al (2012) showed that G6Pase-β expression is important for energy homeostasis in macrophages, and G6pc3−/− macrophages exhibit impairment in respiratory burst, chemotaxis, calcium flux, and phagocytosis activities. Taken together, this suggests that one underlying cause of neutrophil/macrophage dysfunction in GSD-Ib and GSD-Irs is a disturbance in ER energy homeostasis caused by a loss of G6Pase-β/G6PT-mediated glucose/G6P recycling. More recently, the mechanism of neutrophil dysfunction in GSD-Ib was shown to arise from activation of the hypoxia-inducible factor-1α (HIF-1α)/ peroxisome proliferators-activated receptor-γ (PPAR-γ) pathway (Jun et al 2014). The functional coupling of G6Pase-β and G6PT suggests that the HIF-1α/PPAR-γ pathway also plays a role in neutrophil dysfunction in GSD-Irs. However, this pathway remains to be investigated in neutrophils of GSD-Irs patients or mice.
Clinical therapies for GSD-Ia and GSD-Ib
As severe metabolic diseases, both GSD-Ia and GSD-Ib are juvenile lethal if not treated. Presently, metabolic abnormalities of GSD-Ia and GSD-Ib can be adequately controlled with dietary therapy augmented by drug therapy; neutropenia in GSD-Ib can be treated by granulocyte colony-stimulating factor (G-CSF) therapy; and neutropenia with IBD in GSD-Ib can be treated with a combination of G-CSF and 5-aminosalicylic acid (reviewed in Visser et al 2002; Koeberl et al 2009; Chou et al 2010; Shah and O'Dell 2013). However, the underlying pathological process remains uncorrected. As a result, complications include hyperlipidemia, hyperuricemia, lactic acidemia, and hepatomegaly are common, while long-term renal disease and HCA with malignant potential occur in GSD-Ia and GSD-Ib. Complications of splenomegaly and myelodysplasia/acute myeloid leukemia are associated with GSD-Ib patients under G-CSF therapy (Visser et al 2002; Chou et al 2010), although acute myeloid leukemia was not reported in GSD-Irs patients under G-CSF therapy (Banka and Newman 2013).
An alternative treatment for correcting metabolic abnormalities in GSD-I is liver or combined liver/kidney transplantation, while correction of myeloid dysfunctions in GSD-Ib can be addressed by bone marrow transplantation. However, many consider liver transplantation a treatment of last resort and transplantation-related mortality is higher than for most other medical treatments. Current guidelines from the European study on GSD-I recommend liver transplantation in GSD-I patients with un-resectable and dietary unresponsive HCAs, particularly if associated with serious compression or hemorrhage or in the case of transformation into carcinoma (Rake et al 2002). A recent review showed that 80 GSD-I patients (58 GSD-Ia and 22 GSD-Ib) who have received either liver (52 GSD-Ia and 22 GSD-Ib patients) or combined liver-kidney (6 GSD-Ia patients) transplants have regained normal fasting tolerance and shown improved metabolic controls (Boers et al 2014). The six GSD-Ia patients receiving combined liver-kidney transplants also lacked further renal disease. However, the effectiveness of liver only transplantation for either renal disease in GSD-Ia and GSD-Ib, or myeloid dysfunction in GSD-Ib, remains unclear. The complications observed in such transplants are mostly related to transplantation procedures and subsequent immune suppression (Boers et al 2014). Bone marrow transplantation in a GSD-Ib patient manifesting severe IBD and recurrent infections was shown to improve neutrophil function and reduce IBD severity although a mild neutropenia persisted (Pierre et al 2008). While an isolated case, this promising outcome may support further exploration of this approach in addressing severe myeloid complications in GSD-Ib. Despite the transplant guidelines, the liver transplant priority for GSD-I patients, based on the calculated model of end-stage liver disease score is extremely low (Chou et al 2010). Therefore other therapeutic strategies are required.
Gene therapy
Somatic gene therapy is a promising approach, especially for hydrophobic, transmembrane proteins like G6Pase-α, G6Pase-β, and G6PT, where protein replacement therapies are not practical. A variety of gene transfer vectors including adenovirus vectors, helper-dependent adenovirus vectors, and rAAV vectors have been developed using animal models of GSD-Ia or GSD-Ib (reviewed in Chou and Mansfield 2011). rAAV-mediated gene therapy in both mouse and canine models of GSD-Ia has led to long-term correction of hepatic G6Pase-α deficiency with no detectable toxicity (Koeberl et al 2008; Yiu et al 2010; Weinstein et al 2010; Lee et al 2012, 2013). The most promising results come from GSD-Ia studies using rAAV vectors containing the human G6PC promoter/enhancer (GPE), rAAV8-miGPE (originally named rAAV8-G6Pase) (Koeberl et al 2008) and rAAV8-GPE (Yiu et al 2010; Lee et al 2012), rAAV2/8 vectors expressing human G6Pase-α driven either by a 382-bp of the minimal (mi) GPE (rAAV8-miGPE) or the ~3 kb GPE (rAAV8-GPE). In short term (24–26 week) studies, both rAAV8-GPE and rAAV8-miGPE vectors demonstrated efficacy in treating G6pc−/− mice (Koeberl et al 2008; Yiu et al 2010). The rAAV8-miGPE vector also show efficacy in GSD-Ia dog enabling the treated dogs survived to age 11 months with levels of hepatic G6Pase activity and fasting levels of blood glucose/lactate similar to those of the carrier dogs (Koeberl et al 2008). Metabolic normalization and prolonged correction of fasting hypoglycemia in the GSD-Ia dog was also achieved by dosing the GSD-Ia dog first with rAAV8-CBA-G6Pase, a rAAV8 vector expressing human G6Pase-α directed by the chicken β-actin promoter/CMV enhancer (CBA) then 5 months later re-dosing with rAAV1-CBA-G6Pase (Weinstein et al 2010). The need for second infusion may result from a cell-mediated immune response of hepatic CD8+ lymphocyte infiltration observed primarily in mice infused with a vector containing the CBA/CMA promoter/enhancer (Yiu et al 2010). However, this study demonstrates the efficacy of re-administering a rAAV vector, packaged with a new AAV serotype.
In GSD-Ia mouse studies, direct comparison of rAAV8-GPE and rAAV8-miGPE vectors show that the rAAV8-GPE vector directs significantly higher levels of hepatic G6Pase-α expression, achieves greater reduction in hepatic glycogen accumulation, and leads to a better tolerance of fasting, than the rAAV8-miGPE vector (Lee et al 2013), suggesting that the rAAV8-GPE vector is the best vector to take forward into clinical trials. In a long-term dose-ranging study, Lee et al (2012) further showed that rAAV8-GPE-mediated gene transfer, deliberately titrated down to determine the minimum therapeutic dose, showed that restoring ≥ 3 % of normal hepatic G6Pase-α activity in G6pc−/− mice, was sufficient to maintain glucose homeostasis. The treated mice displayed normal hepatic fat storage, normal blood metabolite and glucose tolerance profiles, reduced fasting blood insulin levels, and had no evidence of hepatic abnormalities or HCA. Fasting hypoglycemia is the hallmark of GSD-Ia and GSD-Ib. It is promising that the rAAV8-GPE-treated mice were able to sustain a 24-hour fast, which is a stress test of the ability of the liver to maintain blood normoglycemia through glycogenolysis and gluconeogenesis catalyzed by the G6Pase-α/G6PT complex in the absence of dietary glucose (Fig. 4). This correlated with an increase in hepatic G6PT mRNA expression and a corresponding increase in microsomal G6P uptake activity, leading to the production of low but sufficient glucose to maintain interprandial glucose homeostasis (Fig. 4).

Pathways for G6P metabolism in the livers of normal, GSD-Ia, and rAAV8-GPE-G6PC-treated GSD-Ia mice during fasting. Shown is a simplified cell containing an enlargement of the ER. During fasting, G6P, the end product of gluconeogenesis and glycogenolysis, is transported from the cytoplasm into the lumen of the ER by G6PT. Inside the ER, G6P is hydrolyzed by G6Pase-α and the resulting glucose transported back into the cytoplasm then released into the circulation. In the GSD-Ia liver, which lacks a functional G6Pase-α, ER-localized G6P cannot be converted to glucose, leading to hypoglycemia following a short fast. The rAAV8-GPE-G6PC-treated-GSD-Ia (rAAV-GSD-Ia) liver, which expresses reduced levels of G6Pase-α but increased levels of G6PT compared to the normal liver, can generate reduced levels of endogenous glucose and maintain interprandial glucose homeostasis. The GLUT2 transporter, responsible for the transport of glucose in and out of the cell, is shown embedded in the plasma membrane. G6PT and G6Pase-α are shown embedded in the ER membrane
Correction of renal disease in GSD-Ia and GSD-Ib has been less extensively studied. The rAAV8-mediated gene transfer results in little or no renal G6Pase-α expression and the abnormal renal pathology persists. This was attributed to poor kidney transduction mediated by the AAV2/8 serotype (Chou and Mansfield 2011). Different AAV serotypes have different tissue transduction efficiencies (Michelfelder and Trepel 2009) and more recent data suggest that rAAV2/9 may be the preferred choice for future renal gene delivery (Zincarelli et al 2008; Rocca et al 2014). However, rAAV2/9-mediated transgene expression in the kidney is still significantly lower than that in the liver. Rocca et al (2014) showed that rAAV2/9 kidney transduction could be improved using a retrograde renal vein injection of the virus in mice. While this may hold future promise for GSD-Ia, a single viral therapy may be difficult to develop at present for GSD-Ib and GSD-Irs, since neither vector targets hematopoietic stem cells effectively. Identification of viral serotypes that effectively transduce all affected tissue types remains one avenue to be explored further. It is also important to keep in mind that serotypes can have very different targeting efficiencies in different species. Only a small number of serotypes have been used in clinical trials to date and there is a need to understand more about the primate specificity of the many serotypes that appear promising in rodents.
G-CSF therapy
The use of G-CSF has improved neutropenia and neutrophil function in GSD-Ib (Visser et al 2002) and GSD-Irs (Boztug et al 2009; Jun et al 2010; McDermott et al 2010) but the underlying mechanism is unclear. Jun et al (2011) undertook studies to elucidate the potential mechanism underlying G-CSF correction of murine G6pc3−/− neutropenia and neutrophil dysfunction. They showed that G-CSF improves neutropenia by increasing neutrophil survival. Moreover, G6pc3−/− mice receiving in vivo G-CSF therapy exhibit normalized neutrophil energy homeostasis and improved functionality evidenced by increased neutrophil glucose uptake and elevated intracellular levels of G6P, lactate, and ATP.
Conclusions
Disorders of the G6Pase/G6PT complex can manifest as GSD-Ia (G6Pase-α deficiency), GSD-Ib (G6PT deficiency), or GSD-Irs (G6Pase-β deficiency). The metabolic abnormalities in GSD-Ia and GSD-Ib are currently treated by dietary therapies which can maintain euglycemia and remove the early symptomatic signs of the disease, but leave the patient vulnerable to chronic complications, including hyperlipidemia, hyperuricemia, hypercalciuria, hypocitraturia, lactic acidemia, along with severe long-term complications of renal disease and HCA. Neutropenia, presenting in GSD-Ib and GSD-Irs is caused by enhanced neutrophil apoptosis, and neutrophil dysfunction as a result of impaired energy homeostasis. G-CSF therapy improves neutropenia by enhancing neutrophil survival and rectifies impaired function by normalizing energy homeostasis. The effective use of gene therapies to correct the disease in animal models of GSD-Ia is very promising with efforts to initiate clinical trials on the horizon. Gene therapy approaches that address just the metabolic deficiencies for GSD-Ib are also promising, but strategies that address both the metabolic and myeloid complications may require a dual vector approach. Gene therapy studies for GSD-Irs have not yet been initiated, but should benefit from the studies of myeloid correction in GSD-Ib.
References
Annabi B, Hiraiwa H, Mansfield BC et al (1998) The gene for glycogen-storage disease type 1b maps to chromosome 11q23. Am J Hum Genet 62(2):400–405
Banka S, Newman WG (2013) A clinical and molecular review of ubiquitous glucose-6-phosphatase deficiency caused by G6PC3 mutations. Orphanet J Rare Dis 8:84
Boers SJ, Visser G, Smit PG, Fuchs SA (2014) Liver transplantation in glycogen storage disease type I. Orphanet J Rare Dis 9(1):47
Boztug K, Appaswamy G, Ashikov A et al (2009) A syndrome with congenital neutropenia and mutations in G6PC3. N Engl J Med 360(1):32–43
Chen LY, Shieh JJ, Lin B et al (2003) Impaired glucose homeostasis, neutrophil trafficking and function in mice lacking the glucose-6-phosphate transporter. Hum Mol Genet 12(19):2547–2558
Chen SY, Pan CJ, Nandigama K, Mansfield BC, Ambudkar SV, Chou JY (2008) The glucose-6-phosphate transporter is a phosphate-linked antiporter deficient in glycogen storage disease type Ib and Ic. FASEB J 22(7):2206–2213
Cheung YY, Kim SY, Yiu WH et al (2007) Impaired neutrophil activity and increased susceptibility to bacterial infection in mice lacking glucose-6-phosphatase-beta. J Clin Invest 117(3):784–793
Chou JY, Mansfield BC (2008) Mutations in the glucose-6-phosphatase-alpha (G6PC) gene that cause type Ia glycogen storage disease. Hum Mutat 29(7):921–930
Chou JY, Mansfield BC (2011) Recombinant AAV-directed gene therapy for type I glycogen storage diseases. Expert Opin Biol Ther 11(8):1011–1024
Chou JY, Mansfield BC (2014) The SLC37 family of sugar-phosphate/phosphate exchangers. Curr Top Membr 73:357–382
Chou JY, Matern D, Mansfield BC, Chen YT (2002) Type I glycogen storage diseases: disorders of the glucose-6-phosphatase complex. Curr Mol Med 2(2):121–143
Chou JY, Jun HS, Mansfield BC (2010) Glycogen storage disease type I and G6Pase-beta deficiency: etiology and therapy. Nat Rev Endocrinol 6(12):676–688
Ekstein J, Rubin BY, Anderson SL et al (2004) Mutation frequencies for glycogen storage disease Ia in the Ashkenazi Jewish population. Am J Med Genet A 129A(2):162–164
Franco LM, Krishnamurthy V, Bali D et al (2005) Hepatocellular carcinoma in glycogen storage disease type Ia: a case series. J Inherit Metab Dis 28(2):153–162
Gerin I, Veiga-da-Cunha M, Achouri Y, Collet JF, Van Schaftingen E (1997) Sequence of a putative glucose 6-phosphate translocase, mutated in glycogen storage disease type Ib. FEBS Lett 419(2–3):235–238
Ghosh A, Shieh JJ, Pan CJ, Sun MS, Chou JY (2002) The catalytic center of glucose-6-phosphatase. HIS176 is the nucleophile forming the phosphohistidine-enzyme intermediate during catalysis. J Biol Chem 277(36):32837–32842
Ghosh A, Shieh JJ, Pan CJ, Chou JY (2004) Histidine 167 is the phosphate acceptor in glucose-6-phosphatase-beta forming a phosphohistidine enzyme intermediate during catalysis. J Biol Chem 279(13):12479–12483
Hiraiwa H, Pan CJ, Lin B, Moses SW, Chou JY (1999) Inactivation of the glucose 6-phosphate transporter causes glycogen storage disease type 1b. J Biol Chem 274(9):5532–5536
Jun HS, Lee YM, Cheung YY et al (2010) Lack of glucose recycling between endoplasmic reticulum and cytoplasm underlies cellular dysfunction in glucose-6-phosphatase-beta-deficient neutrophils in a congenital neutropenia syndrome. Blood 116(15):2783–2792
Jun HS, Lee YM, Song KD, Mansfield BC, Chou JY (2011) G-CSF improves murine G6PC3-deficient neutrophil function by modulating apoptosis and energy homeostasis. Blood 117(14):3881–3892
Jun HS, Cheung YY, Lee YM, Mansfield BC, Chou JY (2012) Glucose-6-phosphatase-beta, implicated in a congenital neutropenia syndrome, is essential for macrophage energy homeostasis and functionality. Blood 119(17):4047–4055
Jun HS, Weinstein DA, Lee YM, Mansfield BC, Chou JY (2014) Molecular mechanisms of neutrophil dysfunction in glycogen storage disease type Ib. Blood in press
Kilpatrick L, Garty BZ, Lundquist KF et al (1990) Impaired metabolic function and signaling defects in phagocytic cells in glycogen storage disease type 1b. J Clin Invest 86(1):196–202
Kim SY, Jun HS, Mead PA, Mansfield BC, Chou JY (2008) Neutrophil stress and apoptosis underlie myeloid dysfunction in glycogen storage disease type Ib. Blood 111(12):5704–5711
Koeberl DD, Pinto C, Sun B et al. (2008) AAV vector-mediated reversal of hypoglycemia in canine and murine glycogen storage disease type Ia. Mol Ther 16(4):665–672
Kishnani PS, Faulkner E, VanCamp S et al (2001) Canine model and genomic structural organization of glycogen storage disease type Ia (GSD Ia). Vet Pathol 38(1):83–91
Koeberl DD, Kishnani PS, Bali D, Chen YT (2009) Emerging therapies for glycogen storage disease type I. Trends Endocrinol Metab 20:252–258
Kuijpers TW, Maianski NA, Tool AT et al (2003) Apoptotic neutrophils in the circulation of patients with glycogen storage disease type 1b (GSD1b). Blood 101(12):5021–5024
Lee YM, Jun HS, Pan CJ et al (2012) Prevention of hepatocellular adenoma and correction of metabolic abnormalities in murine glycogen storage disease type Ia by gene therapy. Hepatology 56(5):1719–1729
Lee YM, Pan CJ, Koeberl DD, Mansfield BC, Chou JY (2013) The upstream enhancer elements of the G6PC promoter are critical for optimal G6PC expression in murine glycogen storage disease type Ia. Mol Genet Metab 110(3):275–280
Lei KJ, Shelly LL, Pan CJ, Sidbury JB, Chou JY (1993) Mutations in the glucose-6-phosphatase gene that cause glycogen-storage-disease type-1a. Science 262(5133):580–583
Lei KJ, Chen H, Pan CJ et al (1996) Glucose-6-phosphatase dependent substrate transport in the glycogen storage disease type-1a mouse. Nat Genet 13(2):203–209
Lin B, Pan CJ, Chou JY (2000) Human variant glucose-6-phosphate transporter is active in microsomal transport. Hum Genet 107(5):526–529
Martin CC, Oeser JK, Svitek CA, Hunter SI, Hutton JC, O'Brien RM (2002) Identification and characterization of a human cDNA and gene encoding a ubiquitously expressed glucose-6-phosphatase catalytic subunit-related protein. J Mol Endocrinol 29(2):205–222
Matern D, Seydewitz HH, Bali D, Lang C, Chen YT (2002) Glycogen storage disease type I: diagnosis and phenotype/genotype correlation. Eur J Pediatr 161(Suppl 1):S10–S19
McCawley LJ, Korchak HM, Cutilli JR et al (1993) Interferon-gamma corrects the respiratory burst defect in vitro in monocyte-derived macrophages from glycogen storage disease type 1b patients. Pediatr Res 34(3):265–269
McDermott DH, De Ravin SS, Jun HS et al (2010) Severe congenital neutropenia resulting from G6PC3 deficiency with increased neutrophil CXCR4 expression and myelokathexis. Blood 116(15):2793–2802
Michelfelder S, Trepel M (2009) Adeno-associated viral vectors and their redirection to cell-type specific receptors. Adv Genet 67:29–60
Mutel E, Abdul-Wahed A, Ramamonjisoa N et al (2011) Targeted deletion of liver glucose-6 phosphatase mimics glycogen storage disease type 1a including development of multiple adenomas. J Hepatol 54(3):529–537
Pan CJ, Lei KJ, Annabi B, Hemrika W, Chou JY (1998) Transmembrane topology of glucose-6-phosphatase. J Biol Chem 273(11):6144–6148
Pan CJ, Lin B, Chou JY (1999) Transmembrane topology of human glucose 6-phosphate transporter. J Biol Chem 274(20):13865–13869
Peng WT, Pan CJ, Lee EJ, Westphal H, Chou JY (2009) Generation of mice with a conditional allele for G6pc. Genesis 47(9):590–594
Pierre G, Chakupurakal G, McKiernan P et al (2008) Bone marrow transplantation in glycogen storage disease type 1b. J Pediatr 152(2):286–288
Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP (2002) Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr 161(Suppl 1):S20–S34
Rocca CJ, Ur SN, Harrison F, Cherqui S (2014) rAAV9 combined with renal vein injection is optimal for kidney-targeted gene delivery: conclusion of a comparative study. Gene Ther 21(6):618–628
Shah KK, O'Dell SD (2013) Effect of dietary interventions in the maintenance of normoglycaemia in glycogen storage disease type 1a: a systematic review and meta-analysis. J Hum Nutr Diet 26(4):329–339
Shieh JJ, Terzioglu M, Hiraiwa H et al (2002) The molecular basis of glycogen storage disease type 1a: structure and function analysis of mutations in glucose-6-phosphatase. J Biol Chem 277(7):5047–5053
Shieh JJ, Pan CJ, Mansfield BC, Chou JY (2003) A glucose-6-phosphate hydrolase, widely expressed outside the liver, can explain age-dependent resolution of hypoglycemia in glycogen storage disease type Ia. J Biol Chem 278(47):47098–47103
Visser G, Rake JP, Labrune P et al (2002) Granulocyte colony-stimulating factor in glycogen storage disease type 1b. Results of the European Study on Glycogen Storage Disease Type 1. Eur J Pediatr 161(Suppl 1):S83–S87
Weinstein DA, Correia CE, Conlon T et al (2010) Adeno-associated virus-mediated correction of a canine model of glycogen storage disease type Ia. Hum Gene Ther 21(7):903–910
Yiu WH, Lee YM, Peng WT et al (2010) Complete normalization of hepatic G6PC deficiency in murine glycogen storage disease type Ia using gene therapy. Mol Ther 18(6):1076–1084
Zincarelli C, Soltys S, Rengo G, Rabinowitz JE (2008) Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther 16(6):1073–1080
Acknowledgement
This research was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health.
Compliance with ethics guidelines
ᅟ
Conflict of Interest
Janice Y. Chou, Hyun Sik Jun, and Brian C. Mansfield declare that they have no conflict of interest.
Informed Consent
This article does not contain any studies with human subjects performed by any of the authors.
Animal Rights
All institutional and national guidelines for the care and use of laboratory animals were followed.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Alberto B Burlina
Rights and permissions
About this article

Cite this article
Chou, J.Y., Jun, H.S. & Mansfield, B.C. Type I glycogen storage diseases: disorders of the glucose-6-phosphatase/glucose-6-phosphate transporter complexes. J Inherit Metab Dis 38, 511–519 (2015). https://doi.org/10.1007/s10545-014-9772-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-014-9772-x