
Abstract
Cobalamin C (Cbl-C) defect is the most common inborn cobalamin metabolism error; it causes impaired conversion of dietary vitamin B12 into its two metabolically active forms, methylcobalamin and adenosylcobalamin. Cbl-C defect causes the accumulation of methylmalonic acid and homocysteine and decreased methionine synthesis. The gene responsible for the Cbl-C defect has been recently identified, and more than 40 mutations have been reported. MMACHC gene is located on chromosome 1p and catalyzes the reductive decyanation of CNCbl. Cbl-C patients present with a heterogeneous clinical picture and, based on their age at onset, can be categorized into two distinct clinical forms. Early-onset patients, presenting symptoms within the first year, show a multisystem disease with severe neurological, ocular, haematological, renal, gastrointestinal, cardiac, and pulmonary manifestations. Late-onset patients present a milder clinical phenotype with acute or slowly progressive neurological symptoms and behavioral disturbances. To improve clinical course and metabolic abnormalities, treatment of Cbl-C defect usually consists of a combined approach that utilizes vitamin B12 to increase intracellular cobalamin and to maximize deficient enzyme activities, betaine to provide a substrate for the conversion of homocysteine into methionine through betaine-homocysteine methyltransferase, and folic acid to enhance remethylation pathway. No proven efficacy has been demonstrated for carnitine and dietary protein restriction. Despite these measures, the long-term follow-up is unsatisfactory especially in patients with early onset, with frequent progression of neurological and ocular impairment. The unfavorable outcome suggests that better understanding of the pathophysiology of the disease is needed to improve treatment protocols and to develop new therapeutic approaches.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cobalamin C (Cbl-C) defect is a panethnic disease and is the most common inborn error of cobalamin metabolism. In a recent study based on the results of 5 years of expanded newborn screening in New York State, its incidence has been estimated as approximately 1:100,000 live births (Weisfeld-Adams et al. 2010). Cbl-C defect causes the impaired conversion of dietary cobalamin into its two metabolically active forms, methylcobalamin (MeCbl) and adenosylcobalamin (AdoCbl). MeCbl is the cofactor for methionine synthase, which catalyzes the conversion of homocysteine into methionine in the cytosol; AdoCbl is the cofactor for methylmalonyl-CoA mutase, which converts methylmalonyl-CoA into succinyl-CoA in mitochondria. The impaired activity of these two enzymes results in accumulation of homocysteine and methylmalonic acid (MMA) as well as in reduced synthesis of methionine. The key investigations for the diagnosis are measurements of plasma amino acid levels and urinary organic acids. The recent introduction of acylcarnitine profiling by tandem mass spectrometry demonstrated that most Cbl-C patients show increased propionylcarnitine in blood (Weisfeld-Adams et al. 2010).
The gene responsible for the Cbl-C defect was identified in 2006 (Lerner-Ellis et al. 2006) as MMACHC. MMACHC gene is located on chromosome 1p and codifies the synthesis of CNCbl decyanase, which catalyzes the decyanation reaction of CNCbl by using reducing equivalents given by cytosolic diflavin oxidoreductase, the prerequisite for CNCbl conversion into the active cofactors (Kim et al. 2008). Recently, it has been suggested that MMACHC protein also has an alkyltransferase activity that catalyzes the dealkylation of newly internalized methylcobalamin and 5′-deoxyadenosylcobalamin, the naturally occurring alkylcobalamins that are present in the diet, in a reaction requiring glutathione transferase activity (Kim et al. 2009;Hannibal et al. 2009).
Clinical features
Cbl-C defect is difficult to diagnose on a clinical basis due to its heterogeneous picture. The severity of presentation can vary considerably, ranging from cases with onset in the neonatal period or in early infancy, to late-presenting ones. Based on the age at onset in a cohort of 50 Cbl-C patients, two distinct phenotypes have been recognized (Rosenblatt et al. 1997). Table 1 summarizes the relevant clinical signs according to the age of onset. Patients presenting symptoms in the first year of life are defined as early onset, whereas those exhibiting clinical signs in childhood or later are defined as late onset. Although there are some clinical similarities between the two groups, patients with late onset show a milder phenotype with better survival and less severe neurological impairment.
Early-onset form
The clinical features of the early-onset form include a multisystem disease with neurological, ocular, hematological, renal, gastrointestinal, cardiac, and pulmonary manifestations. Compared with the classical forms of methylmalonic aciduria (Deodato et al. 2006), the clinical picture of Cbl-C defect is usually less acute. Patients present with feeding difficulties, failure to thrive, somnolence/lethargy, and hypotonia. Minor facial anomalies, such as long face; high forehead; large, flappy, and low-set ears; and a flat filtrum have been reported (Cerone et al. 1999). Given the wide variety of unspecific symptoms, diagnosis is often delayed.
The neurological findings are severe and include hypotonia, developmental delay, microcephaly, seizures, hydrocephalus, and MRI abnormalities. Epilepsy is frequent and is characterized by partial seizures, both simple and complex, sometimes leading to convulsive status epilepticus, with a nonspecific EEG pattern (Biancheri et al. 2002). The MRI changes are characterized in the early disease stage by tetraventricular hydrocephalus and diffuse supratentorial white matter swelling, followed by variable degrees of brain atrophy and white matter abnormalities. Basal ganglia lesions have also been reported (Longo et al. 2005).
Ocular abnormalities are variable and include visual inattention, nystagmus, or wandering ocular movements. Optic atrophy and visual abnormalities appear to be inversely related to the age at onset, with the most pronounced impairment observed in the youngest patients, as well as in those showing basal ganglia involvement at MRI (Patton et al. 2000; Tsina et al. 2005; Ricci et al. 2005). Patients may also develop progressive retinal disease, ranging from subtle retinal nerve fiber layer loss to advanced macular and optic atrophy with salt and pepper pigmentation (Robb et al. 1984; Mitchell et al. 1986; Traboulsi et al. 1992; Gerth et al. 2008). ERG studies indicate that early in life the photopic and scotopic responses follow the lower limits of a normal developmental curve, progressing to attenuated or nonrecordable ERGs, confirming retinal degeneration (Schimel and Mets 2006).
Hemolytic uremic syndrome (HUS), defined as the triad of azotemia, thrombocytopenia, and microangiopathic hemolytic anemia, may characteristically occur in Cbl-C defect (Baumgartner et al. 1979; Geraghty et al. 1992; Russo et al. 1992; Andrès et al. 2006; Sharma et al. 2007). Other cases present primary glomerular disease as segmental glomerulosclerosis or atypical glomerulopathy with some features similar to idiopathic membranoproliferative glomerulonephritis and thrombotic microangiopathy (Brunelli et al. 2002).
Nondegenerative megaloblastic anemia, hypersegmented neutrophilis, thrombocytopenia, or severe pancytopenia are the most common hematological findings (Rosenblatt et al. 1997). A single report describes a patient presenting with macrophage activating syndrome (Wu et al. 2005).
Gastrointestinal involvement manifests with vomiting, glossitis, stomatitis, atrophic gastritis, and protein-loosing enteropathy (Ellaway et al. 1998; Russo et al. 1992).
Cardiopulmonary signs are increasingly observed in Cbl-C patients. Congenital heart diseases include ventricular septal defect, pulmonary stenosis, dysplastic pulmonary valve, atrial defects, and mitral valve prolapse (Andersson et al. 1999; Heinemann et al. 2001; Tomaske et al. 2001; Profitlich et al. 2009). Cardiomyopathy and left ventricular noncompaction have been reported as well (Ogier de Baulny et al. 1998; Longo et al. 2005; De Bie et al. 2009; Profitlich et al. 2009). Other descriptions have reported infants with bronchiolitis-like symptoms, rapid deterioration, and death from cor pulmonale with dilated right ventricle. Postmortem examination revealed pulmonary thromboembolism in the absence of evident cardiac pathology (Brandstetter et al. 1990; Profitlich et al. 2009).
Late-onset form
The late-onset form of the disease is rarer than early-onset (Thauvin-Robinet et al. 2008); patients can present at any time from childhood to adulthood and they can be easily misdiagnosed or missed. Along with milder or no hematological abnormalities, the clinical course is characterized by behavioral and psychiatric disturbances, rapid mental deterioration with confusion and disorientation, dementia, delirium, and psychosis.
Late-onset disease can also present with purely neurological manifestations characterized by extrapyramidal symptoms and gait abnormalities occurring acutely or showing a slowly progressive and/or relapsing-remitting course simulating multiple sclerosis (Gold et al. 1996; Powers et al. 2001; Roze et al. 2003; Bodamer et al. 2001; Tsai et al. 2007; Shinnar and Singer 1984; Thauvin-Robinet et al. 2008). Mielopathic signs represent the clinical expression of a subacute degeneration of spinal cord, characterized by multifocal demyelination with vacuolation of dorsal and lateral columns (Smith et al. 2006), changes similar to classical adult spinal cord degeneration due to vitamin B12 deficiency (Scalabrino et al. 2007; Maamar et al. 2008). Brain MRI findings are less specific and include periventricular white matter abnormalities, cortical atrophy, and bilateral ventricular dilatation (Roze et al. 2003).
Some patients have renal damage in the form of chronic thrombotic microangiopathic glomerulo-nephropathy, which leads to end-stage renal failure (Van Hove et al. 2002). The ocular abnormalities show better prognosis with no definite evidence of retinal degeneration (Gerth et al. 2008). Marfanoid features such as increased arm span, arachnodactyly, joint hyperlaxity, and scoliosis have also been reported (Heil et al. 2007). The characteristic occurrence in late-onset cases of thromboembolic events, mostly localized in the great pulmonary vessels, and of spinal cord degeneration might indicate an age-dependent mechanism. Interestingly, the recent introduction of expanded newborn screening allowed the detection of an asymptomatic affected woman whose newborn baby presented with transient metabolite abnormalities (Lin et al. 2009).
Genetic findings
Since MMACHC gene discovery, mutations have been identified in over 300 patients. The most common genetic abnormality is the c.271dupA, which causes a frameshift at codon 91 and a premature termination at codon 105, accounting for more of the 40% of mutant alleles (Morel et al. 2006; Nogueira et al. 2008). Genotype-phenotype correlations have been attempted. Homozygosity for the c.271dupA and for the c.331 C>T mutations are almost exclusively observed in early-onset cases, whereas patients homozygous for the c.394 C>T mutation usually belong to the late-onset group.
Treatment
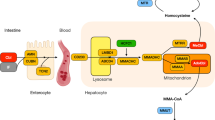
To improve clinical course and metabolic abnormalities, treatment of Cbl-C deficiency is based on a combined approach consisting of supplementation of vitamin B12, betaine, and folic acid. Pharmacological doses of vitamin B12, preferably in the form of hydroxycobalamin (OH-Cbl), are given to maximize enzyme activity. Betaine and folic acid are used to reduce homocysteine and to increase methionine levels (Fig. 1). Betaine provides the substrate for betaine-homocysteine methyltransferase (BHMT), an alternative route for the synthesis of methionine in liver, whereas folic acid enhances the remethylation pathway. Carnitine deficiency, requiring supplementation, can sometimes be observed either due to its loss in buffering intramitochondrial accumulation of MMA or because of reduced synthesis due to low methionine availability. No proven efficacy has been demonstrated with dietary protein restriction. Methionine supplementation has been utilized only very rarely (Smith et al. 2006).

Overview of interrelation of metabolism of cobalamin, homocysteine, and folate. Cyanocobalamin is intracellularly converted into (1) adenosylcobalamin (Adocbl), the cofactor of the methylmalonyl-CoA mutase, which converts methylmalonyl-CoA into succinyl-CoA coenzyme and (2) methylcobalamin (Mecbl), the cofactor of methionine synthase in the remethylation of homocysteine. Homocysteine is a sulfur-containing amino acid derived from the metabolism of methionine. Methionine is adenylated by methionine adenosyltransferase (MAT) into S-adenosylmethionine (SAM), the methyl donor for most methylation reactions. After donating a methyl group, SAM is converted to S-adenosylhomocysteine (SAH), which is hydrolyzed to homocysteine by S-adenosylhomocysteine hydrolase (SAHH). Although this reaction is reversible, the metabolic flow under normal physiological conditions proceeds in the hydrolytic direction, when homocysteine is rapidly removed and concentrations remain low. Homocysteine may be transulfurated to cysteine by cystathionine beta synthase (CBS) or remethylated to methionine via the cobalamin-dependent enzyme methionine synthase (MS). The methylation of homocysteine depends on folate availability and normal activity of the enzyme methylenetetrahydrofolate reductase (MTFHR). An alternative route for the synthesis of methionine is via the betaine-homocysteine methyltransferase (BHMT) reaction, which does not require cobalamin or folate. BHMT activity is lacking in the central nervous system, therefore MTHF is the only methyl donor involved in the methylation of homocysteine to methionine in the brain
Given the known inhibiting effect of nitrous oxide on methionine synthase with direct toxicity at the level of spinal cord (Scott et al. 1981), its use for anesthesia should strongly be contraindicated in patients with Cbl-C defect. This recommendation is further supported by the report of neurological deterioration and death in a child with MTHFR deficiency who underwent anesthesia with nitrous oxide (Selzer et al. 2003).
Some reports emphasize the ineffectiveness of cyanocobalamin both in vivo and in vitro, recommending the use of OH-Cbl (Andersson and Shapira. 1998). So far, only a few studies have investigated, in most cases anecdotally, the biochemical response to OH-Cbl in patients with Cbl-C defect. Bartholomew and co-workers (Bartholomew et al. 1988) reported mild elevation of methylmalonate in one patient, and no significant changes in plasma homocysteine and methionine levels when daily injection of 1 mg OH-Cbl was discontinued for 3 weeks. In a second patient, homocysteine increased within 1 month when OH-Cbl therapy was changed from i.m. to peroral administration. Although most clinicians treating this disorder consider vitamin B12 therapy ineffective when administered perorally, there is evidence that this method of administration may be as effective as i.m. injections in obtaining short-term hematological and neurological response in vitamin B12-deficient patients (Vidal-Alaball et al. 2005; Butler et al. 2006). However, other studies in patients with Cbl-C defect have documented an improvement of metabolic parameters using very high doses, up to 20 mg/day, of parenteral OH-Cbl (Van Hove et al. 2002; Carrillo-Carrasco et al. 2009). It is therefore likely that to obtain a pharmacological effect by OH-Cbl in intracellular cobalamin disorders, the doses needed are much higher than those used to correct low blood levels of vitamin B12.
These reports confirm the lack of formal guidelines for vitamin B12 therapy and do not provide a clear answer as to whether the variable responses seen in patients are primarily related to cobalamin concentration or if other determinants, such as MMACHC genotype, can influence treatment efficacy.
An additional example of the lack of consensus on the optimum treatment is provided by recent reports that indicate as first-line therapy dietary protein restriction combined with daily parenteral OH-Cbl injection, limiting the use of betanine only to those patients with Cbl-C showing persistent hyperhomocysteinemia (Weisfeld-Adams et al. 2010; Profitlich et al. 2009).
Outcome
Despite therapeutic measures, sometimes started soon after initial presentation or even prenatally, the long-term outcome is often unsatisfactory (Andersson et al. 1999; Patton et al. 2000; Huemer et al. 2005; Profitlich et al. 2009; Weisfeld-Adams et al. 2010). Biochemical abnormalities usually improve under treatment but without reaching a complete normalization. Homocysteine levels remain well above normal in the majority of cases (Rosenblatt et al. 1997). The prognosis seems to be worse in the early-onset group, with a high percentage of death in the largest series of patients reported so far (Rosenblatt et al. 1997). In early-onset patients, treatment usually results in improvement of visceral and hematological signs, but in most cases has less efficacy on the neurological outcome and on ocular symptoms (Rosenblatt et al. 1997; Andersson et al. 1999; Tsina et al. 2005; Smith et al. 2006; Profitlich et al. 2009).Degrees of neurological and cognitive impairment are almost invariably present, regardless of the age at diagnosis or treatment initiation (Rosenblatt et al. 1997; Andersson et al. 1999). The course of retinal degeneration, which sometimes can lead to blindness, appears to be unaltered (Tsina et al. 2005; Gerth et al. 2008). Also in late-onset cases, despite some improvement with B12 therapy, neurological recovery is rare and incomplete (Thauvin-Robinet et al. 2008). In contrast to early-onset cases, patients with a late disease onset usually show less severe ocular involvement with no definite evidence of retinal degeneration (Gerth et al. 2008).
Pathophysiology
An exact comprehensive understanding of the pathophysiology of Cbl-C defect has still not been achieved, and it is likely that the synergistic effect of different mechanisms, which include the accumulation of putatively toxic metabolites and the deficiency of missed products downstream of the enzymatic defect(s), is responsible for the multisystem organ involvement.
Methylmalonate toxicity
Although the levels of MMA in Cbl-C patients are usually lower than those observed in “classical” forms of methylmalonic aciduria (Fowler et al. 2008), accumulation of this compound may actively contribute to neurological dysfunction during attacks of acute encephalopathy but could also act as a chronic neurotoxic substance. Methylmalonate and its related compounds (e.g., methylcitrate, malonic acid, and propionyl-COA) may impair mitochondrial energy metabolism in the central nervous system (CNS) and also induce secondary excitotoxic cell damage (Kölker et al. 2008). More recently, a new neuropathogenetic mechanism, the “dicarboxylic acid trapping hypothesis,” was proposed after demonstration that within the brain compartment there is a strong accumulation of dicarboxylic acids and a weak clearing capacity of blood brain barrier for these compounds (Kölker et al. 2008). According to this theory, methylmalonate would be preferentially stored in CNS, further contributing to toxic neurological damage. MMA could also play a role as a possible nephrotoxin, causing proteinuria and renal tubular injury or impairing transport processes of the renal proximal tubule (Sauer et al. 2009).
Homocysteine toxicity
A significant amount of evidence suggests that homocysteine presents toxic effects in multiple ways, affecting different biological systems. Besides Cbl-C defect, all inherited diseases causing severe homocysteine elevation are associated with cognitive and neurological impairment. Furthermore, it is now well known that increased homocysteine concentrations as well as folate and vitamin B12 deficiencies are associated with cognitive deficit, dementia, poorer neurocognitive performance, and Alzheimer’s and Parkinson’s diseases (McCaddon and Kelly 1992; Regland et al. 1992; Yasui et al. 2000; Smith 2008). The various pathomechanisms proposed to explain the role of homocysteine in brain and cognitive functions include the involvement of cerebrovascular circulation through an ischemia-reperfusion mechanism, causing Alzheimer-type pathology, white matter damage and silent infarctions; direct neurotoxicity causing cell death; the initiation of a cellular cascade of apoptosis and hyperphosphorylation of tau- and beta-amyloid; interference with DNA repair system; stimulation of endoplasmatic reticulum stress response; and increased expression of APP or its hydrolysis to amyloid B through a hypomethylation process (Outinen et al. 1999; Kruman et al. 2000; Mattson and Shea 2003; Wong et al. 2006; Obeid and Herrmann 2006).
It was the original case of Cbl-C defect that presented early in life with widespread vascular lesions that led to the elucidation of the homocysteine theory of atherosclerosis (McCully 1992). Hyperhomocysteinemia was then confirmed to be a risk factor for atherosclerotic vascular disease (Welch and Loscalzo 1998). In contrast, patients with Down’s syndrome seem to be protected against atherosclerosis due to overexpression of the genes that cause diminished plasma homocysteine levels (Licastro et al. 2006). Although it has been proved that high homocysteine levels are associated with an increased risk of cardiovascular diseases, a recent meta-analysis of published trials suggests that there is no evidence that homocysteine-lowering interventions prevent vascular events or reduce mortality risk (Martì-Carvajal et al. 2009). The effects of homocysteine in inducing vascular damage include increased oxidative stress through formation of reactive oxygen species, the up-regulation of prothrombotic factors XII and V, the stimulation of proinflammatory pathways and of lipid peroxidation, and the stimulation and proliferation of vascular smooth muscle cells (Wang et al. 2000; Nowak-Göttl et al. 2003; Weiss 2005; Zou and Banerjee 2005; Papatheodorou and Weiss 2007)
On these bases, it is evident that homocysteine contributes to cause many of the clinical symptoms observed in Cbl-C defect.
Methionine deficiency
In contrast to CBS deficiency, in which hyperhomocysteinemia is accompanied by elevation of methionine and in which the vascular system involvement is usually characterized by thromboembolic attacks mostly localized in great venous vessels, in inherited conditions causing hyperhomocysteinemia and hypomethioninemia (i.e., Cbl-C, CblD, CblE, CblG, and MTHFR deficiency), the vascular system appears to be mostly affected at the level of small arterial vessels. Remarkably, HUS and hydrocephalus, which represent characteristic signs of Cbl-C defect, and other conditions with hyperhomocysteinemia and hypomethioninemia have never been reported in CBS deficiency (Mudd et al. 1985). Diffuse microangiopathy with intimal proliferation causes HUS and, according to most of the recent hydrodynamic theories, also causes communicating hydrocephalus (Geraghty et al. 1992; Rossi et al. 2001; Greitz 2004). On these bases, it may be hypothesized that, in addition to the pathogenetic role of hyperhomocysteinemia in the vascular system, the localization and type of vascular damage could depend on other factors, and in particular on methionine levels.
Numerous studies support a relevant role of methionine also in CNS function. Neuronal cells cultured in methionine-deficient medium exhibit the highest levels of homocysteine-induced apoptosis; demyelination and cognitive decline can be related to methionine and SAM deficiency; and over-expression of CBS in Down’s syndrome causes methionine and SAM deficiencies (Pogribna et al. 2001; Kruman et al. 2002; Tchantchou et al. 2006). Furthermore, methionine deficiency has been proposed as one of the mechanisms causing dementia in Alzheimer’s disease and AIDS (Regland and Gottfries 1992; Smith and Greenwood 2008). Reduced serum and CSF methionine and SAM levels have been reported in AIDS-associated myelopathy (Di Rocco et al. 2002) with white matter vacuolization similar to Cbl-C spinal degeneration. Interestingly, demyelination is associated with reduced SAM levels in cerebrospinal fluid of patients with inborn errors of the methyl-transfer pathway, a process that can be reversed by increasing SAM availability with oral methionine treatment (Surtees et al. 1991). Methionine deficiency may therefore result in inadequate methylation of various essential compounds, and homocysteine and SAH excess can further deteriorate this condition because of their potent inhibitory effect on methyltransferase enzymes. Methylation is necessary for the synthesis of nucleic acids and neurotransmitters, regulates gene expression, and modifies protein function (Detich et al. 2003). Since a perturbation of DNA and histone methylation, especially during periods of organogenesis, has important effects on gene expression and cardiac development (Robertson 2002), this mechanism has been hypothesized to be the underlying cause of cardiopathy in Cbl-C patients (Profitlich et al. 2009). Furthermore, the creatine biosynthetic pathway also depends on the availability of labile methyl groups, and it has been proposed that creatine deficiency and guanidinoacetate elevation can contribute to the neurological phenotype in Cbl-C patients (Bodamer et al. 2005). However, this finding was not confirmed by other studies reporting normal plasma levels of guanidinoacetate with borderline normal creatine levels as well as normal brain creatine as detectable by proton magnetic spectroscopy (Longo et al. 2005; Younessi et al. 2009).
Conclusion
Understanding the pathophysiology and improving treatment in patients with Cbl-C defect still remains a great challenge. The unfavorable outcome observed in most patients confirms that actual interventions, mostly focused on improving biochemical parameters, are not sufficient to prevent organ damage and that individual differences may also influence the response to therapies. Furthermore, the majority of treatment-related studies report anecdotal results on small series of patients. At present, there is no unifying and generally accepted concept for the pathogenesis of Cbl-C defect and, despite substantial evidence suggesting a possible role of methionine and of methylation reactions as contributing factors, this route has not so far been fully investigated (Smith et al. 2006). From this point of view, the systematic use of methionine supplementation could represent an important innovation for the treatment of Cbl-C defect that needs future multicenter clinical trials to evaluate its efficacy.
Abbreviations
- AdoCbl:
-
Adenosylcobalamin
- BHMT:
-
Betaine-homocysteine methyltransferase
- Cbl:
-
Cobalamin
- CBS:
-
Cystathionine beta synthase
- HUS:
-
Hemolytic uremic syndrome
- MeCbl:
-
Methylcobalamin
- MMA:
-
Methylmalonic acid
- MTHFR:
-
Methyl tetrahydrofolate reductase
- OH-Cbl:
-
Hydroxycobalamin
- SAM:
-
S-adenosylmethionine
- SAH:
-
S-adenosylhomocysteine
References
Andersson HC, Shapira E (1998) Biochemical and clinical response to hydroxycobalamin versus cyanocobalamin treatment in patients with methylmalonic academia and homocystinuria (Cbl-C). J Pediatr 132:121–124
Andersson HC, Marble M, Shapira E (1999) Long-term outcome in treated combined methylmalonic acidemia and homocystinemia. Genet Med 1(4):146–150
Andrès E, Affenberger S, Federici L, Korganow AS (2006) Pseudo-thrombotic microangiopathy related to cobalamin deficiency. Am J Med 119(12):e3
Bartholomew DW, Batshaw ML, Allen RH et al (1988) Therapeutic approaches to cobalamin-C methylmalonic acidemia and homocystinuria. J Pediatr 112(1):32–39
Baumgartner ER, Wick H, Maurer R, Egli N, Steinmann B (1979) Congenital defect in intracellular cobalamin metabolism resulting in homocysteinuria and methylmalonic aciduria. I. Case report and histopathology. Helv Paediatr Acta 34(5):465–482
Biancheri R, Cerone R, Rossi A et al (2002) Early-onset cobalamin C/D deficiency: epilepsy and electroencephalographic features. Epilepsia 43(6):616–622
Bodamer OA, Rosenblatt DS, Appel SH, Beaudet AL (2001) Adult-onset combined methylmalonic aciduria and homocystinuria (Cbl-C). Neurology 56(8):1113
Bodamer OA, Sahoo T, Beaudet AL et al (2005) Creatine metabolism in combined methylmalonic aciduria and homocystinuria. Ann Neurol 57(4):557–560
Brandstetter Y, Weinhouse E, Splaingard ML, Tang TT (1990) Cor pulmonale as a complication of methylmalonic acidemia and homocystinuria (Cbl-C type). Am J Med Genet 36(2):167–171
Brunelli SM, Meyers KE, Guttenberg M, Kaplan P, Kaplan BS (2002) Cobalamin C deficiency complicated by an atypical glomerulopathy. Pediatr Nephrol 17(10):800–803
Butler CC, Vidal-Alaball J, Cannings-John R et al (2006) Oral vitamin B12 versus intramuscular vitamin B12 for vitamin B12 deficiency: a systematic review of randomized controlled trials. Fam Pract 23(3):279–285
Carrillo-Carrasco N, Sloan J, Valle D, Hamosh A, Venditti CP (2009) Hydroxocobalamin dose escalation improves metabolic control in Cbl-C. J Inherit Metab Dis 32(6):728–731
Cerone R, Schiaffino MC, Caruso U, Lupino S, Gatti R (1999) Minor facial anomalies in combined methylmalonic aciduria and homocystinuria due to a defect in cobalamin metabolism. J Inherit Metab Dis 22(3):247–250
De Bie I, Nizard SD, Mitchell GA (2009) Fetal dilated cardiomyopathy: an unsuspected presentation of methylmalonic aciduria and hyperhomocystinuria, Cbl-C type. Prenat Diagn 29(3):266–270
Deodato F, Boenzi S, Santorelli FM, Dionisi-Vici C (2006) Methylmalonic and propionic aciduria. Am J Med Genet C Semin Med Genet 142C(2):104–112
Detich N, Hamm S, Just G, Knox JD, Szyf M (2003) The methyl donor S-adenosylmethionine inhibits active demethylation of DNA: a candidate novel mechanism for the pharmacological effects of S-adenosylmethionine. J Biol Chem 278(23):20812–20820
Di Rocco A, Bottiglieri T, Werner P et al (2002) Abnormal cobalamin-dependent transmethylation in AIDS-associated myelopathy. Neurology 58(5):730–735
Ellaway C, Christodoulou J, Kamath R, Carpenter K, Wilcken B (1998) The association of protein-losing enteropathy with cobalamin C defect. J Inherit Metab Dis 21(1):17–22
Fowler B, Leonard JV, Baumgartner MR (2008) Causes of and diagnostic approach to methylmalonic acidurias. J Inherit Metab Dis 31(3):350–360
Geraghty MT, Perlman EJ, Martin LS et al (1992) Cobalamin C defect associated with hemolytic-uremic syndrome. J Pediatr 120(6):934–937
Gerth C, Morel CF, Feigenbaum A, Levin AV (2008) Ocular phenotype in patients with methylmalonic aciduria and homocystinuria, cobalamin C type. J AAPOS 12(6):591–596
Gold R, Bogdahn U, Kappos L et al (1996) Hereditary defect of cobalamin metabolism (homocystinuria and methylmalonic aciduria) of juvenile onset. J Neurol Neurosurg Psychiatr 60(1):107–108
Greitz D (2004) The hydrodynamic hypothesis versus the bulk flow hypothesis. Neurosurg Rev 27(4):299–300
Hannibal L, Kim J, Brasch NE et al (2009) Processing of alkylcobalamins in mammalian cells: a role for the MMACHC (cbl-C) gene product. Mol Genet Metab 97(4):260–266
Heil SG, Hogeveen M, Kluijtmans LAJ, van Dijken PJ, van de Berg GB, Blom HJ, Morava E (2007) Marfanoid features in a child with combined methylmalonic aciduria and homocystinuria (CblC type). J Inherit Metab Dis 30:811
Heinemann MK, Tomaske M, Trefz FK, Bosk A, Baden W, Ziemer G (2001) Ventricular septal defect closure in a neonate with combined methylmalonic aciduria/homocystinuria. Ann Thorac Surg 72(4):1391–1392
Huemer M, Simma B, Fowler B, Suormala T, Bodamer OA, Sass JO (2005) Prenatal and postnatal treatment in cobalamin C defect. J Pediatr 147(4):469–472
Kim J, Gherasim C, Banerjee R (2008) Decyanation of vitamin B12 by a trafficking chaperone. Proc Natl Acad Sci USA 105(38):14551–14554
Kim J, Hannibal L, Gherasim C, Jacobsen DW, Banerjee R (2009) A human vitamin B12 trafficking protein uses glutathione transferase activity for processing alkylcobalamins. J Biol Chem 284(48):33418–33424
Kölker S, Sauer SW, Hoffmann GF, Müller I, Morath MA, Okun JG (2008) Pathogenesis of CNS involvement in disorders of amino and organic acid metabolism. J Inherit Metab Dis 31:194–204
Kruman II, Culmsee C, Chan SL et al (2000) Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J Neurosci 20(18):6920–6926
Kruman II, Kumaravel TS, Lohani A et al (2002) Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. J Neurosci 22(5):1752–1762
Lerner-Ellis JP, Tirone JC, Pawelek PD et al (2006) Identification of the gene responsible for methylmalonic aciduria and homocystinuria, Cbl-C type. Nat Genet 38(1):93–100
Licastro F, Marocchi A, Penco S et al (2006) Does Down’s syndrome support the homocysteine theory of atherogenesis? Experience in elderly subjects with trisomy 21. Arch Gerontol Geriatr 43(3):381–387
Lin HJ, Neidich JA, Salazar D et al (2009) Asymptomatic maternal combined homocystinuria and methylmalonic aciduria (Cbl-C) detected through low carnitine levels on newborn screening. J Pediatr 155(6):924–927
Longo D, Fariello G, Dionisi-Vici C et al (2005) MRI and 1H-MRS findings in early-onset cobalamin C/D defect. Neuropediatrics 36(6):366–372
Maamar M, Mezalek ZT, Harmouche H, Adnaoui M, Aouni M, Maaouni A (2008) Contribution of spinal MRI for unsuspected cobalamin deficiency in isolated sub-acute combined degeneration. Eur J Intern Med 19(2):143–145
Martì-Carvajal AJ, Solà I, Lathyris D, Salanti G (2009) Homocysteine lowering interventions for preventing cardiovascular events. Cochrane Database Syst Rev. 7(4):CD006612
Mattson MP, Shea TB (2003) Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci 26(3):137–146
McCaddon A, Kelly CL (1992) Alzheimer’s disease: a ‘cobalaminergic’ hypothesis. Med Hypotheses 37(3):161–165
McCully KS (1992) Homocystinuria, arteriosclerosis, methylmalonic aciduria, and methyltransferase deficiency: a key case revisited. Nutr Rev 50(1):7–12
Mitchell GA, Watkins D, Melançon SB et al (1986) Clinical heterogeneity in cobalamin C variant of combined homocystinuria and methylmalonic aciduria. J Pediatr 108(3):410–415
Morel CF, Lerner-Ellis JP, Rosenblatt DS (2006) Combined methylmalonic aciduria and homocystinuria (Cbl-C): phenotype-genotype correlations and ethnic-specific observations. Mol Genet Metab 88(4):315–321
Mudd SH, Skovby F, Levy HL et al (1985) The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am J Hum Genet 37(1):1–31
Nogueira C, Aiello C, Cerone R et al (2008) Spectrum of MMACHC mutations in Italian and Portuguese patients with combined methylmalonic aciduria and homocystinuria, Cbl-C type. Mol Genet Metab 93(4):475–480
Nowak-Göttl U, Duering C, Kempf-Bielack B, Sträter R (2003) Thromboembolic diseases in neonates and children. Pathophysiol Haemost Thromb 33(5–6):269–274
Obeid R, Herrmann W (2006) Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett 580(13):2994–3005
Ogier de Baulny H, Gérard M, Saudubray JM, Zittoun J (1998) Remethylation defects: guidelines for clinical diagnosis and treatment. Eur J Pediatr 157(Suppl 2):S77–S83
Outinen PA, Sood SK, Pfeifer SI et al (1999) Homocysteine-induced endoplasmic reticulum stress and growth arrest leads to specific changes in gene expression in human vascular endothelial cells. Blood 94(3):959–967
Papatheodorou L, Weiss N (2007) Vascular oxidant stress and inflammation in hyperhomocysteinemia. Antioxid Redox Signal 9(11):1941–1945
Patton N, Beatty S, Lloyd IC, Wraith JE (2000) Optic atrophy in association with cobalamin C (Cbl-C) disease. Ophthalmic Genet 21(3):151–154
Pogribna M, Melnyk S, Pogribny I, Chango A, Yi P, James SJ (2001) Homocysteine metabolism in children with Down syndrome: in vitro modulation. Am J Hum Genet 69(1):88–95
Powers JM, Rosenblatt DS, Schmidt RE et al (2001) Neurological and neuropathologic heterogeneity in two brothers with cobalamin C deficiency. Ann Neurol 49(3):396–400
Profitlich LE, Kirmse B, Wasserstein MP, Diaz GA, Srivastava S (2009) High prevalence of structural heart disease in children with Cbl-C-type methylmalonic aciduria and homocystinuria. Mol Genet Metab 98(4):344–348
Regland B, Gottfries CG (1992) Slowed synthesis of DNA and methionine is a pathogenetic mechanism common to dementia in Down’s syndrome, AIDS and Alzheimer’s disease? Med Hypotheses 38(1):11–19
Regland B, Abrahamsson L, Blennow K, Gottfries CG, Wallin A (1992) Vitamin B12 in CSF: reduced CSF/serum B12 ratio in demented men. Acta Neurol Scand 85(4):276–281
Ricci D, Pane M, Deodato F et al (2005) Assessment of visual function in children with methylmalonic aciduria and homocystinuria. Neuropediatrics 36(3):181–185
Robb RM, Dowton SB, Fulton AB, Levy HL (1984) Retinal degeneration in vitamin B12 disorder associated with methylmalonic aciduria and sulfur amino acid abnormalities. Am J Ophthalmol 97(6):691–696
Robertson KD (2002) DNA methylation and chromatin. Unraveling the tangled web. Oncogene 21(35):5361–5379
Rosenblatt DS, Aspler AL, Shevell MI, Pletcher BA, Fenton WA, Seashore MR (1997) Clinical heterogeneity and prognosis in combined methylmalonic aciduria and homocystinuria (Cbl-C). J Inherit Metab Dis 20(4):528–538
Rossi A, Biancheri R, Tortori-Donati P (2001) The pathogenesis of hydrocephalus in inborn errors of the single carbon transfer pathway. Neuropediatrics 32(6):335–336
Roze E, Gervais D, Demeret S et al (2003) Neuropsychiatric disturbances in presumed late-onset cobalamin C disease. Arch Neurol 60(10):1457–1462
Russo P, Doyon J, Sonsino E, Ogier H, Saudubray JM (1992) A congenital anomaly of vitamin B12 metabolism: a study of three cases. Hum Pathol 23(5):504–512
Sauer SW, Opp S, Haarmann A, Okun JG, Kölker S, Morath MA (2009) Long-term exposure of human proximal tubule cells to hydroxycobalamin[c-lactam] as a possible model to study renal disease in methylmalonic acidurias. J Inherit Metab Dis 32(6):720–727, Epub 2009 Oct 10
Scalabrino G, Veber D, Mutti E (2007) New pathogenesis of the cobalamin-deficient neuropathy. Med Secoli 19(1):9–18
Schimel AM, Mets MB (2006) The natural history of retinal degeneration in association with cobalamin C (cbl C) disease. Ophthalmic Genet 27(1):9–14
Scott JM, Dinn JJ, Wilson P, Weir DG (1981) Pathogenesis of subacute combined degeneration: a result of methyl group deficiency. Lancet 2(8242):334–337
Selzer RR, Rosenblatt DS, Laxova R, Hogan K (2003) Adverse effect of nitrous oxide in a child with 5, 10-methylenetetrahydrofolate reductase deficiency. N Engl J Med 349(1):45–50
Sharma AP, Greenberg CR, Prasad AN, Prasad C (2007) Hemolytic uremic syndrome (HUS) secondary to cobalamin C (Cbl-C) disorder. Pediatr Nephrol 22(12):2097–2103
Shinnar S, Singer HS (1984) Cobalamin C mutation (methylmalonic aciduria and homocystinuria) in adolescence. A treatable cause of dementia and myelopathy. N Engl J Med 311(7):451–454
Smith AD (2008) The worldwide challenge of the dementias: a role for B vitamins and homocysteine? Food Nutr Bull 29(Suppl 2):S143–S172
Smith KL, Greenwood CE (2008) Weight loss and nutritional considerations in Alzheimer disease. J Nutr Elder 27(3–4):381–403
Smith SE, Kinney HC, Swoboda KJ, Levy HL (2006) Subacute combined degeneration of the spinal cord in Cbl-C disorder despite treatment with B12. Mol Genet Metab 88(2):138–145
Surtees R, Leonard J, Austin S (1991) Association of demyelination with deficiency of cerebrospinal-fluid S-adenosylmethionine in inborn errors of methyl-transfer pathway. Lancet 338(8782–8783):1550–1554
Tchantchou F, Graves M, Ortiz D, Chan A, Rogers E, Shea TB (2006) S-adenosyl methionine: a connection between nutritional and genetic risk factors for neurodegeneration in Alzheimer’s disease. J Nutr Health Aging 10(6):541–544
Thauvin-Robinet C, Roze E, Couvreur G et al (2008) The adolescent and adult form of cobalamin C disease: clinical and molecular spectrum. J Neurol Neurosurg Psychiatry 79(6):725–728, Epub 2008 Feb 1
Tomaske M, Bosk A, Heinemann MK et al (2001) Cbl-C/D defect combined with haemodynamically highly relevant VSD. J Inherit Metab Dis 24(4):511–512
Traboulsi EI, Silva JC, Geraghty MT, Maumenee IH, Valle D, Green WR (1992) Ocular histopathologic characteristics of cobalamin C type vitamin B12 defect with methylmalonic aciduria and homocystinuria. Am J Ophthalmol 113(3):269–280
Tsai AC, Morel CF, Scharer G et al (2007) Late-onset combined homocystinuria and methylmalonic aciduria (Cbl-C) and neuropsychiatric disturbance. Am J Med Genet A 143A(20):2430–2434
Tsina EK, Marsden DL, Hansen RM, Fulton AB (2005) Maculopathy and retinal degeneration in cobalamin C methylmalonic aciduria and homocystinuria. Arch Ophthalmol 123(8):1143–1146
Van Hove JL, Van Damme-Lombaerts R, Grünewald S et al (2002) Am J Med Genet 111(2):195–201
Vidal-Alaball J, Butler CC, Cannings-John R, et al (2005) Oral vitamin B12 versus intramuscular vitamin B12 for vitamin B12 deficiency. Cochrane Database Syst Rev 20(3):CD004655
Wang G, Siow YL, K O (2000) Homocysteine stimulates nuclear factor kappaB activity and monocyte chemoattractant protein-1 expression in vascular smooth-muscle cells: a possible role for protein kinase C. Biochem J 352(Pt 3):817–826
Weisfeld-Adams JD, Morrissey MA, Kirmse BM et al (2010) Newborn screening and early biochemical follow-up in combined methylmalonic aciduria and homocystinuria, Cbl-C type, and utility of methionine as a secondary screening analyte. Mol Genet Metab 99(2):116–123
Weiss N (2005) Mechanisms of increased vascular oxidant stress in hyperhomocysteinemia and its impact on endothelial function. Curr Drug Metab 6(1):27–36
Welch GN, Loscalzo J (1998) Homocysteine and atherothrombosis. N Engl J Med 338(15):1042–1050
Wong A, Mok V, Fan YH, Lam WW, Liang KS, Wong KS (2006) Hyperhomocysteinemia is associated with volumetric white matter change in patients with small vessel disease. J Neurol 253(4):441–447
Wu S, Gonzalez-Gomez I, Coates T, Yano S (2005) Cobalamin C disease presenting with hemophagocytic lymphohistiocytosis. Pediatr Hematol Oncol 22(8):717–721
Yasui K, Kowa H, Nakaso K, Takeshima T, Nakashima K (2000) Plasma homocysteine and MTHFR C677T genotype in levodopa-treated patients with PD. Neurology 55(3):437–440
Younessi D, Moseley K, Yano S (2009) Creatine metabolism in combined methylmalonic aciduria and homocystinuria disease revisited. Ann Neurol 65(4):481–482
Zou CG, Banerjee R (2005) Homocysteine and redox signaling. Antioxid Redox Signal 7(5–6):547–559
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Brian Fowler
References to electronic databases: Methylmalonic aciduria and homocystinuria cbl-C type: OMIM #277400
MMACHC gene: OMIM *609831
Competing interest: None declared.
Rights and permissions
About this article
Cite this article
Martinelli, D., Deodato, F. & Dionisi-Vici, C. Cobalamin C defect: natural history, pathophysiology, and treatment. J Inherit Metab Dis 34, 127–135 (2011). https://doi.org/10.1007/s10545-010-9161-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-010-9161-z