
Summary
There are few reports of congenital disorders of glycosylation (CDGs) in the Asian population, although they have been reported worldwide. We identified a Malaysian infant female at 2 days of life with CDG type Ia. The diagnosis was suspected on the basis of inverted nipples and abnormal fat distribution. She had cerebellar hypoplasia and developed coagulopathy, hypothyroidism and severe pericardial effusion and died at 7 months of life. The diagnosis was supported by abnormal serum transferrin isoform pattern that showed elevated levels of the disialotransferrin isoform and trace levels of the asialotransferrin isoform. Enzyme testing of peripheral leukocytes showed decreased level of phosphomannomutase (PMM) activity (0.6 nmol/min per mg protein, normal range 1.6–6.2) and a normal level of phosphomannose isomerase activity (19 nmol/min per mg protein, normal range 12–25), indicating a diagnosis of CDG type Ia. Mutation study of the PMM2 gene showed the patient was heterozygous for both the common p.R141H (c.422T>A) mutation and a novel sequence change in exon 7, c.618C>A. The latter change is predicted to result in the replacement of the highly conserved phenylalanine residue at position 206 with a leucine residue (p.F206L) and occurs in the same codon as the previously reported p.F206S mutation. Analysis of 100 control chromosomes has shown that the p.F206L sequence change is not present, making it highly likely that this change is functionally important. To the best of our knowledge, this is the first report of CDG in the Malay population. Prenatal diagnosis was successfully performed in a subsequent pregnancy for this family.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Congenital disorders of glycosylation (CDGs) are multisystem disorders caused by defects in the synthesis of the glycan moiety of glycoproteins or other glycoconjugates (Jaeken 2003). CDG type I is caused by defects of N-glycan assembly, whereas CDG type II is caused by defects of N-glycan processing. The list of disease-causing defects is increasing, with the most common defect being due to the deficiency of phosphomannomutase (PMM) leading to CDG Ia (OMIM #212065). CDG is a multisystem disease with a wide range of clinical presentation. The phenotype can range from very mild to very severe with two broad groups of presentation: those with neurological disease and those with multivisceral disease. There is considerable overlap between these groups and the clinical diagnosis of CDG during the neonatal period requires a high index of suspicion. While CDG has been reported in patients world-wide, there are few reports of this condition in the Asian population (Enns et al 2002; Kondo et al 1999; Newell et al 2003; Westphal et al 2001).
Case report
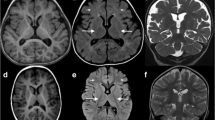
The patient was a 1-day-old Malay baby girl who was referred for dysmorphic features. The parents were second cousins and there was no significant family history of note. The antenatal period was uneventful and the baby was delivered at term via lower-segment Caesarean section with a birth weight of 3.6 kg, length of 46 cm and head circumference of 38 cm. During the postnatal examination, she was noted to have bilateral inverted nipples, and abnormal fat distribution over her thighs, buttocks and suprapubic regions. A flat nasal bridge and prominent nares were also noted (Figs. 1, 2, 3). The rest of the examination, including neurological findings, was unremarkable. CDG type I was suspected.

Dysmorphic features

Bilateral inverted nipples

Abnormal fat distribution
Cerebellar hypoplasia was noted on cranial magnetic resonance imaging. Chest radiography showed cardiomegaly and echocardiography showed pericardial effusion. Serum transferrin isoform analysis showed significantly elevated levels of the disialotransferrin isoform and trace levels of the asialotransferrin isoform. This pattern is typical for CDG type I.
Enzyme testing of peripheral leukocytes showed a markedly decreased level of PMM activity (0.6 nmol/min per mg protein, normal range 1.6–6.2) and a normal level of phosphomannose isomerase activity (19 nmol/min per mg protein, normal range 12–25), indicating a diagnosis of CDG type Ia. Mutation study of the PMM2 gene (accession no. NM_000303.1) was performed and showed that the patient was heterozygous for both the common p.R141H (c.422T>A) mutation and a novel sequence change in exon 7, c.618C>A, which is predicted to result in the amino acid change p.F206L. Analysis of 100 control chromosomes has shown that the p.F206L change is not present. Further, a cross-species comparison of the PMM protein sequence has shown that this phenylalanine residue is conserved from slime mould to primates (Fig. 4) and is, therefore, likely to be functionally important. Analysis of parental DNA samples indicated that p.R141H was maternally inherited, while p.F206L was paternally inherited.

Cross-species comparison of the PMM protein sequence shows that the phenylalanine residue is conserved from slime mould to primates
Further investigations showed impaired coagulation with prolonged prothrombin time and partial thromboplastin time, hypothyroidism and persistent hypoalbuminaemia. The patient was treated with anti-cardiac failure therapy, l-thyroxine and intravenous albumin infusion. She presented with recurrent episodes of acute respiratory distress as a result of the severe pericardial effusion. She was transfused with fresh frozen plasma before pericardiocentesis could be performed. The pericardial fluid was sterile. She succumbed to her illness at the age of 7 months.
The couple had prenatal diagnosis performed for the second pregnancy. Chorionic villus sampling (CVS) was performed at 11 weeks’ gestation. PMM2 mutation study on the CVS showed the presence of the p.R141H mutation only, indicating that the fetus was a carrier for CDG type Ia. A healthy baby was delivered at term. Postnatal study confirmed the findings of the prenatal diagnosis. Peripheral blood leukocytes of the newborn baby’s cord blood showed a level of PMM activity at the lower end of the normal range (1.6 nmol/min per mg protein, normal 1.6–6.2), which is consistent with the carrier status determined by prenatal testing. Postnatal reviews of the child at 6 and at 12 months of life showed normal developmental milestones.
Discussion
Reports of CDG are uncommon in Asian patients. To the best of our knowledge, this is the first report of CDG in the Malay population. It is possible that this condition may be underdiagnosed owing to the low awareness of this disorder among healthcare staff. To achieve an early clinical diagnosis, a high index of suspicion based on recognition of the diverse clinical features is required. Early investigations such as the use of serum transferrin isoform analysis are helpful in achieving the diagnosis. The diagnosis of CDG type Ia can then be confirmed by measurement of PMM activity in either peripheral blood leukocytes or cultured skin fibroblasts. Further investigations such as cranial magnetic resonance imaging and echocardiography can assist in detecting the structural abnormalities often seen in CDG type Ia. Other major complications such as coagulopathy and endocrine deficiencies should be sought, as these are treatable.
The diagnosis of CDG type Ia assisted in the process of genetic counselling. While there is no definitive curative therapy for this disorder at the moment, other supportive care options were available. Mutation studies of the PMM2 gene in our patient identified a common mutation p.R141H and a novel mutation p.F206L. The p.R141H is a known severe mutation and is reported to account for 40% of disease alleles in CDG Ia patients (Marquardt and Denecke 2003). Patients homozygous for the p.R141H mutation have not been detected, indicating that this genotype leads to fetal death in utero. Although p.F206L has not been reported previously, a second change at this codon (p.F206S) has been reported to be pathogenic (Schollen et al 2002). In addition, the phenylalanine residue at position 206 is highly conserved across a broad range of species, indicating that it is likely that this residue is functionally important. Analysis of unaffected control individuals has shown that the p.F206L change does not occur at high levels within the normal population. We therefore believe it highly likely that p.F206L is the second causative mutation in this patient. The severe clinical features in this patient indicate that p.F206L is a relatively severe mutation. Further studies would be required in order to determine the frequency of both the p.R141H and p.F206L mutations in the Asian population. With the availability of information on the molecular genetics, prenatal diagnosis using chorionic villus sampling at 11 weeks’ gestation was successfully carried out for this family.
Abbreviations
- CDG:
-
congenital disorder of glycosylation
- PMM:
-
phosphomannomutase
References
Enns GM, Steiner RD, Buist N, et al (2002) Clinical and molecular features of congenital disorder of glycosylation in patients with type I sialotransferrin pattern and diverse ethnic origins. J Pediatr 141(5): 695–700. doi:10.1067/mpd.2002.128658
Jaeken J (2003) Congenital disorders of glycosylation (CDG): It’s all in it! J Inherit Metab Dis 26: 99–118. doi:10.1023/A:1024431131208
Kondo I, Muzugizhi K, Yoneda Y, et al (1999) Missense mutations in phosphomannomutase2 gene in two Japanese families with carbohydrate-deficient glycoprotein syndrome type I. Clin Genet 55: 50–54. doi:10.1034/j.1399-0004.1999.550109.x
Marquardt T, Denecke J (2003) Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr 162: 359–379.
Newell JW, Seo NS, Enns GM, McCracken M, Mantovani JF, Freeze HH (2003) Congenital disorder of glycosylation Ic in patients of Indian origin. Mol Genet Metab 79: 221–284. doi:10.1016/S1096-7192(03)00089-1
Schollen E, Martens K, Geuzens E, Matthijs G. (2002) DHPLC analysis as a platform for molecular diagnosis of congenital disorders of glycosylation (CDG). Eur J Hum Genet 10: 643–648. doi:10.1038/sj.ejhg.5200858
Westphal V, Enns GM, McCracken M, Freeze HH (2001) Functional analysis of novel mutations in a congenital disorder of glycosylation Ia patient with mixed Asian ancestry. Mol Genet Metab 73:71–76. doi:10.1006/mgme.2001.3174
Acknowledgement
We acknowledge the assistance of Professor Dr Peng-Chiong Tan, Department of Obstetric and Gynecology, University of Malaya Medical Centre, in performing the prenatal diagnosis. We thank Dr Su-Lin Chow for her clinical contribution.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicating editor: Jaak Jaeken
JIMD Short Report #150 (2009) Online
Competing interests: None declared
References to electronic databases: Congenital disorder of glycosylation 1a: OMIM #212065.
Rights and permissions
About this article
Cite this article
Thong, M.K., Fietz, M., Nicholls, C. et al. Congenital disorder of glycosylation type Ia in a Malaysian family: Clinical outcome and description of a novel PMM2 mutation. J Inherit Metab Dis 32 (Suppl 1), 41–44 (2009). https://doi.org/10.1007/s10545-009-1031-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-009-1031-1