
Abstract
Background
Congenital disorders of glycosylation are rare conditions caused by genetic defects in glycan synthesis, processing or transport. Most congenital disorders of glycosylation involve defects in the formation or transfer of the lipid-linked oligosaccharide precursor of N-linked glycans. SLC35A2-CDG (previously CDG-IIm) is caused by hemizygous or heterozygous mutations in the X-linked gene SLC35A2 that encodes a UDP-galactose transporter. To date there have only been 10 reported patients with SLC35A2 mutations. Importantly, the patient presented here was not identified in infancy by transferrin isoform analysis, the most common testing to identify patients with a congenital disorder of glycosylation.
Case presentation
A 27 month old girl with developmental delay, central hypotonia, cerebral atrophy, and failure to thrive with growth retardation was identified by whole exome sequencing to have a mosaic missense variant in SLC35A2 (c.991G > A). This particular variant has been previously reported in a male as a mutation. Comparison of all clinical findings and new information on growth pattern, growth hormone testing and neurodevelopmental evaluation are detailed on the patient presented.
Conclusion
This patient report increases the clinical and scientific knowledge of SLC35A2-CDG, a rare condition. New information on reduced growth, growth hormone sufficiency, lack of seizures, and neurodevelopmental status are presented. This new information will be helpful to clinicians caring for individuals with SLC35A2-CDG. This report also alerts clinicians that transferrin isoform measurements do not identify all patients with congenital disorders of glycosylation.
Similar content being viewed by others

Background
Congenital disorders of glycosylation (CDGs) are rare hereditary disorders associated with facial dysmorphism and neurological impairments including hypotonia, seizures, intellectual disability, and demyelinating neuropathy [1, 2]. There are two groups: one (CDG-I) that results from defects in glycan addition to the N-terminal; and a second group (CDG-II) that occurs due to defects in the processing of protein-bound glycans or vesicular transport [2, 3]. The most frequent of these is PMM2-CDG which includes 80% of the diagnosed cases [1].
SLC35A2-CDG (previously CDG-IIm) is a rare form of CDG caused by mutations in the X-linked gene SLC35A2 that encodes a UDP-galactose transporter [4,5,6]. In eight patients with SLC35A2-CDG in the literature (Table 1), manifestations include seizures, failure to thrive, delayed myelination and cerebral atrophy [4, 6,7,8]. There are two other reports of SLC35A2 variants in the literature, but no clinical information was provided for these cases, so they will not be discussed further [9, 10]. In this report we describe a female child with growth retardation, hypotonia and global developmental delay who was found by whole exome sequencing (WES) to be mosaic for an SLC35A2 mutation previously reported [4].
Case presentation
A female infant was delivered vaginally after a pregnancy complicated by hyperemesis and preterm labor. Birth weight was 2.89 kg (13th centile) and length was 53 cm (87th centile). At birth, resuscitation was required, followed by neonatal jaundice and feeding difficulties.
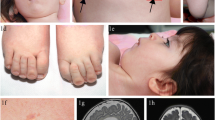
During the first year of life, the patient had central hypotonia, gross motor delay, and failure to thrive. At age 9 months, brain magnetic resonance imaging (MRI) showed delayed myelination pattern on both T1- and T2-weighted images, generalized cerebral atrophy (white matter > grey matter), and mildly prominent extra-axial cerebrospinal fluid spaces consistent with a low brain volume (Fig. 1a-c). An electroencephalogram (EEG) obtained for staring spells was negative for clinical or subclinical seizures. Metabolic testing included normal lactate, pyruvate, free carnitine, CK, acylcarnitine profile, folate, vitamin B12, plasma amino acids, and very long chain fatty acids. Chromosomal microarray was normal. Methylation studies for Prader-Willi syndrome were normal. Transferrin isoforms by immunoaffinity liquid chromatography and electrospray mass spectrometry [11] at 12 months revealed elevated A-oligo/Di-oligo ratio of 0.02 (< 0.011) and Trisialo/Di-oligo ratio of 0.07 (< 0.05). Mono-oligo/Di-oligo, ApoCIII-1/ApoCIII-2, and ApoCIII-0/ApoCIII-2 ratios were within normal range. Repeat transferrin isoforms testing at 13.5 months was normal (Table 2). Due to weight loss and dehydration, a gastrostomy tube was placed at age 2 years. An ophthalmological examination showed esotropia, amblyopia, and hyperopic astigmatism. Otoacoustic emission testing was normal.

Brain MRI. a-c: SLC35A2-CDG patient at 22 months. d-f: Normal age and sex matched patient for comparison. a, d T1-weighted axial images, b, e TurboFLAIR axial images, c, f T2-weighted coronal images. Images a and b demonstrate mild widening of the cerebral sulci that is most notable in the frontal and temporal lobes. Shortening of the head in the anterior-posterior dimension (brachycephaly) was noted, which may be incidental as there are no other findings to suggest craniosynostosis. Image c also demonstrates cerebral volume loss. Delayed myelination is also best seen on c, exhibiting the lack of T2 hypointensity diffusely in the cerebral white matter (compared to f) expected for age. The delayed myelination is also evident on the TurboFLAIR image b as the lack of characteristic hypointensity in the posterior limbs of the internal capsules (long arrows) and in the deep white matter of the frontal lobes (short arrows)
At age 27 months, she was first seen at our institution. Examination revealed a weight of 10.4 kg (3rd centile), length of 78.4 cm (3rd centile) and OFC of 47.8 cm (50th centile). She had a high forehead, thick eyebrows with synophrys, midface hypoplasia, broad philtrum, thick lower lip, short palpebral fissures with deep-set eyes, bilateral epicanthal folds, flat nasal bridge, and a broad tip to the nose (Fig. 2). Other physical findings included prominent calcaneal fat pads anteromedial to the heels and a 2 cm irregular cafe au lait spot of the upper right groin area. A three-generation family history revealed that the patient’s mother was adopted, but the maternal family was of Mexican/European descent. The father’s family was said to be of European descent, but little else was known. There was no concern for consanguinity.

Facial appearance of patient 1 with SLC35A2 mutation
For WES, DNA was extracted from peripheral blood. The Agilent Clinical Research Exome Kit (Agilent, Santa Clara, CA) was used for sequence enrichment and the resulting libraries were sequenced on an Illumina HiSeq 2000 instrument (Illumina, San Diego, CA). Paired 100 base reads were mapped and genotyped using a custom informatics pipeline. The patient was found to have a mosaic variant c.991G > A or p.Val331lle (V331I) in the SLC35A2 gene in 20% of reads (Additional file 1: Figure S1). The mother was negative for the c.991G > A variant in blood. This variant has been previously been reported as a mutation in the mosaic state in a male [4]. X-chromosome inactivation pattern was detected by methylation analysis of the androgen receptor (AR) gene locus by methylation-sensitive restriction digest, PCR and fragment analysis [12]. This revealed non-random skewed inactivation (ratio 85:15), where a ratio greater than 80:20 is considered as skewed inactivation.
Additional imaging studies were performed at 30 months of age including a renal ultrasound, ECG, and echocardiogram, all of which were normal. Other studies included an antithrombin III of 116% (85–135), thrombin time of 16.3 s (13–19), PTT of 34 (22–37 s) and INR of 1.03 (0.86–1.14), which were normal, and protein C of 118% (40–92), which was elevated. Because of her slow growth, IgA and IgG tissue transglutaminase antibodies were tested and both were normal. IgA level was 91 mg/dl (20–160). The liver transaminases and albumin (3.8 g/d (3.4–5) were normal except for mild elevated level of aspartate transaminase (AST) at 65 U/L (0–50).
At 34 months she underwent endocrine evaluation for growth failure. Her length was 81.8 cm (− 3.1 SDS), weight 10.6 kg (− 2.3 SDS) and head circumference was 49.3 cm (+ 0.59 SDS) (Additional file 2: Figure S2 A, B). Her growth hormone provocative stimulation test showed a peak growth hormone (GH) of 21 ng/ml (normal response > 10 ng/ml); insulin-like growth factor 1 (IGF-1) level was 65 ng/ml (16–178, − 0.1 SDS), insulin growth factor binding protein 3 (IGFBP3) was 3.1 μg/ml (0.8–3.9), and acid labile subunit (ALS) was slightly elevated at 11 mg/ml (1.9–10). Free T4 was 1.22 ng/ml (0.76–1.46) and TSH was 1.48 (0.4–4) mU/L.
In terms of neurocognitive development, the patient was delayed in reaching early milestones. She sat alone at age 15 months, crawled at 18 months, and walked independently at 2.5 years. She began speaking words after 2 years of age and started using word combinations at 3.5 years. A neurodevelopmental evaluation at age 53 months was conducted using the Bayley Scales of Infant and Toddler Development, Third Edition. Results indicated impaired performance on cognitive and language testing (19-to 22-month level). Expressive communication consisted primarily of single words and gestures, with some 2-and 3-word phrases. Fine motor function was at a 27-month level and gross motor function was at a 19-month level. The patient was socially engaged, cooperative, and good-natured during testing. Hyperactivity and distractibility were notable. Caregiver ratings of adaptive function were in the range of mild-to-moderate impairment. Although developmental progress was significantly slower than typical, the patient’s history indicated a pattern of consistent incremental gains over time, with beneficial response to early intervention services.
The CARE guidelines were followed in the presentation of this case.
Discussion and conclusions
There have been eight reported cases of SLC35A2-CDG with clinical details, including 2 males and 6 females, all caused by de novo mutations of SLC35A2 [4, 6,7,8]. There is substantial variability in clinical symptoms within the group (Table 1). This variability could be due to the type of mutation (frameshift vs. missense), the level of mosaicism, or the degree of X-chromosome inactivation. Three female patients (our patient, patient 6 and patient 7) were found to have skewed X-inactivation. Kodera et al. found only the wild-type SLC35A2 allele to be expressed in the lymphoblastoid cell lines of patients 6 and 7 predicting that further mutant alleles were silenced by the skewed X-chromosome inactivation [6]. Additionally, Dorre et al. discovered only wild-type alleles in fibroblasts and only mutated alleles in lymphocytes of patient 5 [7]. These findings suggest that complicated patterns of X-chromosome inactivation may play a role in phenotypic severity in SLC35A2-CDG. Additionally, both previously reported male patients (Table 1) were mosaic for a SLC35A2 mutation, suggesting that this disorder may be X-linked dominant and that males must be mosaic for survival.
All reported patients, including the current one, had brain MRI abnormalities (Table 1) and had neurological symptoms ranging from developmental delay and hypotonia to early onset infantile encephalopathy with severe seizures and hypsarrhythmia. Only patient 1 (our patient) and patient 4 [4] who were both mosaic for the c.991G > A mutation in SLC35A2, did not have seizures (Table 1). Microcephaly was present in patients 3 and 4 [4] but not in our patient. Additionally, our patient does not have impaired kidney function, shortened limbs or severe gastrointestinal issues that were present in patient 4 [4].
Our patient demonstrated considerably more advanced neurodevelopmental function than previously described patients, many of whom were nonverbal and unable to walk [6, 7]. The invariable presence of central nervous system (CNS) deficits has led other authors to hypothesize that the negative selection of the mutant SLC35A2 allele seen in most tissues may happen to a lesser degree in the CNS with most of the neurons expressing the mutant SLC35A2 allele [6].
Transferrin isoform testing by mass spectrometry or isoelectric focusing to detect elevated levels of hypoglycosylated serum transferrins is the current practice for diagnosis of N-glycosylation defects [4, 5]. Our patient and four of the 8 other SLC35A2-CDG cases (Table 2) had abnormal transferrin glycosylation profiles before the first year of life, but testing had normalized by ages 1–3 years. A current hypothesis is that during infancy the body selects hepatocytes with the mutant allele [4]. Even in the most common CDG, PMM2-CDG, there have been two cases of normal or nearly normal transferrin levels in children: one child with only slightly abnormal and a second with abnormal transferrin levels at 1 year that normalized by age 8 [13]. Such normalization of transferrin IEF after the first year of life in some children with SLC35A2-CDG emphasizes the importance of performing WES in children with clinical findings compatible with CDG [14] but normal transferrin levels.
One predominant issue in patients with CDG is failure to thrive, which is thought to be caused by a combination of poor nutritional status and/or impairment of the GH-IGF-1 cascade due to hypoglycosylation of growth factors and their receptors. Significantly decreased levels of ALS, IGFBP3, IGF-1 and IGF-2, and ternary complex formation have been described in other patients with PMM2-CDG [15,16,17].
Dhaunsi [18] found low IGF-1 levels in 3 patients with CDG-II (specific mutations not specified) and selective impairment of IGF-1-induced synthesis of DNA in the lymphoblasts of children with either PMM2-CDG or CDG-II compared to controls. Of interest, both children with PMM2-CDG and with CDG-II had remarkable hypoglycosylation of their IGF-1 receptor protein compared to controls, which may explain the decreased IGF-1-induced synthesis of DNA, even in children with normal IGF-1 levels. The IGF-1 receptor is a glycoprotein and requires proper post-translational glycosylation of its protein for proper function [19]. Impaired IGF-1 receptor glycosylation and signaling may explain the growth failure seen in our patient in the presence of normal IGF-1 level and GH production. GH and/or IGF-1 therapies in children with CDG may have a role in improving the growth in children with CDG and growth failure.
This report provides additional information on the phenotypic spectrum of SLC35A2-CDG, a rare condition now described in 9 patients. Because of the rarity of CDG, regardless of type, data on linear growth in children with CDG is limited. This is the first report to evaluate the growth axis of CDG-IIm including a provocative GH stimulation test. Although our patient’s peak GH response was high and her growth hormone dependent IGF-1, IGFBP3, and ALS levels were within normal ranges, her growth and weight gain have continued to be below the 3rd percentile.
In summary, WES testing should be utilized to identify rare CDG disorders in children with normal transferrin levels, but who have other clinical findings that are usually seen in CDG.
Abbreviations
- ALS:
-
Acid labile subunit
- AR:
-
Androgen receptor
- ASH:
-
Asymmetric septal hypertrophy
- CDG:
-
Congenital disorder of glycosylation
- ECG:
-
Electrocardiogram
- EEG:
-
Electroencephalogram
- GERD:
-
Gastroesophageal reflux disease
- GH:
-
Growth hormone
- IGF:
-
Insulin like growth factor
- IGFBP:
-
insulin like growth factor binding protein
- MRI:
-
Magnetic resonance imaging
- ND:
-
Not described
- PCR:
-
Polymerase chain reaction
- TSH:
-
Thyroid stimulating hormone
- VSD:
-
Ventricular septal defect
- WES:
-
Whole exome sequencing
References
Freeze HH, Eklund EA, Ng BG, Patterson MC. Neurology of inherited glycosylation disorders. Lancet Neurol. 2012;11(5):453–66.
Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat. 2009;30(12):1628–41.
Aebi M, Helenius A, Schenk B, Barone R, Fiumara A, Berger EG, Hennet T, Imbach T, Stutz A, Bjursell C, et al. Carbohydrate-deficient glycoprotein syndromes become congenital disorders of glycosylation: an updated nomenclature for CDG. First International Workshop on CDGS. Glycoconj J. 1999;16(11):669–71.
Ng BG, Buckingham KJ, Raymond K, Kircher M, Turner EH, He M, Smith JD, Eroshkin A, Szybowska M, Losfeld ME, et al. Mosaicism of the UDP-galactose transporter SLC35A2 causes a congenital disorder of glycosylation. Am J Hum Genet. 2013;92(4):632–6.
Scott K, Gadomski T, Kozicz T, Morava E. Congenital disorders of glycosylation: new defects and still counting. J Inherit Metab Dis. 2014;37(4):609–17.
Kodera H, Nakamura K, Osaka H, Maegaki Y, Haginoya K, Mizumoto S, Kato M, Okamoto N, Iai M, Kondo Y, et al. De novo mutations in SLC35A2 encoding a UDP-galactose transporter cause early-onset epileptic encephalopathy. Hum Mutat. 2013;34(12):1708–14.
Dorre K, Olczak M, Wada Y, Sosicka P, Gruneberg M, Reunert J, Kurlemann G, Fiedler B, Biskup S, Hortnagel K, et al. A new case of UDP-galactose transporter deficiency (SLC35A2-CDG): molecular basis, clinical phenotype, and therapeutic approach. J Inherit Metab Dis. 2015;38(5):931–40.
Kimizu T, Takahashi Y, Oboshi T, Horino A, Koike T, Yoshitomi S, Mori T, Yamaguchi T, Ikeda H, Okamoto N, et al. A case of early onset epileptic encephalopathy with de novo mutation in SLC35A2: clinical features and treatment for epilepsy. Brain and Development. 2017;39(3):256–60.
Alsubhi S, Alhashem A, Faqeih E, Alfadhel M, Alfaifi A, Altuwaijri W, Alsahli S, Aldhalaan H, Alkuraya FS, Hundallah K, et al. Congenital disorders of glycosylation: the Saudi experience. Am J Med Genet A. 2017;173(10):2614–21.
Al-Shamsi A, Hertecant JL, Souid AK, Al-Jasmi FA. Whole exome sequencing diagnosis of inborn errors of metabolism and other disorders in United Arab Emirates. Orphanet J Rare Dis. 2016;11(1):94.
Lacey JM, Bergen HR, Magera MJ, Naylor S, O'Brien JF. Rapid determination of transferrin isoforms by immunoaffinity liquid chromatography and electrospray mass spectrometry. Clin Chem. 2001;47(3):513–8.
Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet. 1992;51(6):1229–39.
Vermeer S, Kremer HP, Leijten QH, Scheffer H, Matthijs G, Wevers RA, Knoers NA, Morava E, Lefeber DJ. Cerebellar ataxia and congenital disorder of glycosylation Ia (CDG-Ia) with normal routine CDG screening. J Neurol. 2007;254(10):1356–8.
Hennet T. Diseases of glycosylation beyond classical congenital disorders of glycosylation. Biochim Biophys Acta. 2012;1820(9):1306–17.
Kjaergaard S, Muller J, Skovby F. Prepubertal growth in congenital disorder of glycosylation type Ia (CDG-Ia). Arch Dis Child. 2002;87(4):324–7.
Miller BS, Duffy MM, Addo OY, Sarafoglou K. rhIGF-1 therapy for growth failure and IGF-1 deficiency in congenital disorder of glycosylation Ia (PMM2 deficiency). J Investig Med High Impact Case Rep. 2013;1(3):2324709613503316.
Miller BS, Khosravi MJ, Patterson MC, Conover CA. IGF system in children with congenital disorders of glycosylation. Clin Endocrinol. 2009;70(6):892–7.
Dhaunsi GS. Receptor-mediated selective impairment of insulin-like growth factor-1 activity in congenital disorders of glycosylation patients. Pediatr Res. 2016;81(3):526–30.
Siddle K. The insulin receptor and type I IGF receptor: comparison of structure and function. Prog Growth Factor Res. 1992;4(4):301–20.
Acknowledgements
The authors thank the patient and family for assisting us with the manuscript. The authors thank GeneDx, Gaithersburg MD, for providing Additional file 1: Figure S1. The authors thank Patricia A. Buenzle for assistance with the manuscript.
Availability of data and materials
All data generated and analyzed in this study are included in this published article and its supplemental information files.
Author information
Authors and Affiliations
Contributions
KW examined the patient, detailed the laboratory studies, and was a major contributor in writing the manuscript. KS evaluated the growth data and endocrine evaluation results, wrote the section interpreting that data, and edited the manuscript. LCS provided the neurologic information and edited the manuscript. EIP provided the neurodevelopmental information, wrote that section, and edited the manuscript. JS interpreted the genetic testing and edited the manuscript. DN evaluated the radiographic studies, provided the MRI illustrations and edited the manuscript. MB provided and interpreted the WES information and sequencing data for the SLC35A2 mutation. MEP provided much of the patient report information, was a major contributor in writing the manuscript and was the major editor of the manuscript. All authors have read, reviewed, and approved the final manuscript for submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval for a single case report is not required at the authors’ University. We have a letter of confirmation from the IRB of the University of Minnesota that this case study does not meet the federal definition of human subjects’ research and does not require IRB review and approval.
Consent for publication
A consent form has been signed by the parent of the patient in this report that included consent for medical data and an image taken in a clinical setting. This form is available as needed.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Sequencing data for SLC35A2 mutation. [A]- Integrative Genomics Viewer image of next generation sequencing data showing chrX:g.48762195C > T (HG19) variant present in 20% of reads. [B] Bidirectional Sanger sequence confirmation of c.991G > A SLC35A2 variant in peripheral blood sample from proband. [C] Bidirectional Sanger sequence data from maternal blood sample demonstrating absence of the c.991G > A variant. Note- next generation sequence data shown in relation to the HG19 chromosome X reference sequence, while Sanger sequencing data is presented in relation to the SLC35A2 reference transcript (NM_005660.2) which is located on the opposite strand. (TIFF 1521 kb)
Additional file 2:
Figure S2. A, B. A. Length growth over time. B. Weight growth over time. (ZIP 65 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Westenfield, K., Sarafoglou, K., Speltz, L.C. et al. Mosaicism of the UDP-Galactose transporter SLC35A2 in a female causing a congenital disorder of glycosylation: a case report. BMC Med Genet 19, 100 (2018). https://doi.org/10.1186/s12881-018-0617-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-018-0617-6