
Abstract
Since three-dimensional (3D) cell culture models better reflect tissues in vivo in terms of cell shape and microenvironment compared to conventional monolayer cultures, 3D tissue culture substrates gain more importance for a wide range of biological applications like drug discovery, toxicological studies, cancer and stem cell research. In this study we developed a method for the fabrication of 3D cell culture substrates in a multiwell plate format by microstructuring the bottom of 96-well cell culture plates using an ultrasonic embossing process. The resulting microstructured area consists of cubic microcavities in which adherent multicellular aggregates can be formed. We performed the biological evaluation of the system with the liver-derived human cell-line HepG2 and compared the novel substrate with a commercially available 3D culture system comprising porous alginate sponges. Metabolic activity (alamarBlue® reduction) and induction of four biotransformation enzymes (EROD, ECOD, UGT, SULT) were determined by fluorimetry or HPLC. Our results revealed that HepG2 cells in microstructured plates showed a higher mitochondrial activity, as well as enzyme activity of ECOD and UGT after treatment with an inducer when compared to cells cultured in alginate sponges at otherwise comparable conditions. Since we have modified standard cell culture plates, the obtained system is adaptable to automated screening and might be useful for all kinds of cultures including adult, progenitor and stem cells which need a 3D culture configuration to restore or maintain the differentiated status.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The early identification of potential hepatotoxins or toxins in general, during the process of drug development or the accreditation of chemicals necessitates cost-effective, fast and reliable tests for the validation process. In this context, cell-based assay systems represent a helpful tool in drug discovery and toxicological studies, as cellular models exhibit in contrast to subcellular fractions like microsomes, the necessary transporter proteins for drug uptake, nuclear receptors for enzyme induction and the respective detoxification enzymes (Lam and Benet 2004; Soars et al. 2007; Vermeir et al. 2005). Compared to traditional monolayer cell cultures, three-dimensional (3D) culture models better reflect complex systems like tissues in terms of cell shape, as well as microenvironment and are therefore suitable to restore and maintain the differentiated status of adult cells like hepatocytes in vitro (Abu-Absi et al. 2002; Koide et al. 1989; Kunz-Schughart et al. 2004; Wu et al. 1999). For this reason there is an increasing demand on 3D tissue culture substrates for cell-based high-throughput screenings (HTS), especially in pharmaceutical and chemical industries. Commercially available culture systems concerning 3D culture in multiwell cell culture plates comprise mainly sponges (e.g. collagen or calcium phosphate sponges), or hydrogels made of natural polymers like alginate, extracellular matrix components or synthetic peptides hydrogels (reviewed in Justice et al. (2009)). The fabrication process of such porous sponge-like matrices is based on techniques like freeze-drying and gas foaming (no use of a porogen) or solvent casting and particulate leaching (use of a solid porogen) (reviewed in Lee et al. (2008)). Moreover, culture systems using other approaches like microstructured or grooved substrates often comprise microfabrication technologies like soft lithography and photo lithography (Khademhosseini et al. 2006; Park and Lee 2005). Other concepts apply coatings onto the bottom of the wells, for instance a polystyrene solution which is foamed to form microcavities (Cheng et al. 2008).
In contrast to above mentioned techniques, we use a culture system developed at the Karlsruhe Institute of Technology that is based on a microstructured polymer chip that serves as scaffold for the three-dimensional cultivation of cells in micro-bioreactors. This chip has a central grid-like microstructured area with cubic microcavities in which cells can organize into uniform multicellular aggregates (Gottwald et al. 2007; Weibezahn et al. 1993). The cavities of the chip are open to the top and have a side length of 300 μm in each direction (w × l × d). The manufacturing process comprises a microreplication technique, such as microinjection molding or vacuum hot embossing of poly(methyl methacrylate) (PMMA) or polycarbonate to fabricate the container array of the scaffold. In a second step, a solvent-vapor-welding technique is used to bond a porous membrane to the open bottom of the chip. As the geometry of the chip array proved to be suitable for the generation of uniform adherent multicellular aggregates (Altmann et al. 2008; Gottwald et al. 2007; Rieke et al. 2008), our intention was to transfer the chip design to multiwell cell culture plates for HTS applications.
Typically, standard multiwell cell culture plates are fabricated by injection molding. In principle, microstructured multiwell plates may be produced in the same manner using a microstructured molding tool. However, these microstructures would increase the adhesion during the demolding step which might compromise the quality and the production time as well as the costs. For that reason our new concept is to microstructure consecutively the wells of commercially available standard cell culture plates in an additional process especially in the case of prototype or small batch fabrication.
While injection molding requires the polymer to be completely molten and injected into the mold, the additional microstructuring is performed under conditions which avoid any deformation of the frame of the multiwell plate. For that reason embossing technologies at temperatures below the melting point of the polymer have to be applied. Using the hot embossing technology (Worgull 2009) the polymer is heated just above its glass transition temperature and then the mold is pressed into the softened material. Alternatively a heated embossing tool could be pressed into the corresponding cavities of the multiwell cell culture plate. When the wells are microstructured consecutively, it is sufficient to heat the polymer locally. One promising method to generate heat on the surface of a polymer is the use of ultrasonic vibrations. For ultrasonic welding, a well-known technology for different bonding applications, heat is generated by ultrasonic vibrations and the polymer is molten locally. We adapted the ultrasonic welding technology for the fabrication of microfluidic components and tested this method to microstructure a polymer surface (Truckenmüller et al. 2006). Similar developments have been made by other groups who either used ultrasonic embossing (Khuntontong et al. 2009; Liu and Dung 2005; Schomburg et al. 2011) or the ultrasonic micro hot embossing technology (Mekaru et al. 2007) where ultrasonic vibrations support a conventional hot embossing process.
While in many cases a special structured ultrasonic horn is used, we employed a standard ultrasonic welding machine in combination with a special designed embossing tool which acts as an anvil. Thus, instead of the ultrasonic horn only the embossing counter tool has to be replaced to change the design of the microstructures. To avoid the adhesion problems during the demolding step, the microstructuring is done consecutively with a single embossing tool, which also reduces its fabrication costs. Furthermore, it was possible to use the same microstructured embossing tool for different layouts of cell culture plates (e.g. 24-well or 96-well plates or standard Petri dishes).
2 Materials and methods
2.1 Materials
All chemicals, reagents and standards, as well as the protease inhibitor cocktail and propidium iodide were purchased from Sigma-Aldrich (Taufkirchen, Germany) unless otherwise noted. Minimum Essential Medium (MEM), glutamine and trypsin were purchased from Gibco® (Invitrogen, Karlsruhe, Germany), penicillin/streptomycin, sodium pyruvate, non essential amino acids and foetal bovine serum were obtained from PAA Laboratories (Cölbe, Germany). Experiments were performed in 96-well cell culture plates from Greiner Bio-One (Cat.-No. 655180; Frickenhausen, Germany), which were modified by ultrasonic embossing, and in 96-well plates with porous alginate sponges termed AlgiMatrix® 3D Culture System from Gibco® (Invitrogen, Karlsruhe, Germany)
2.2 Fabrication of microstructured multiwell plates for 3D cell culture
2.2.1 Ultrasonic embossing
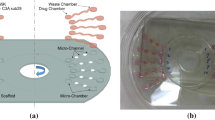
Standard multiwell plates were structured using an ultrasonic embossing process. A schematic view of the ultrasonic embossing system is shown in Fig. 1(a). The main component is the pneumatic ultrasonic welding system Herrmann PS DIALOG digital control 1000 from Herrmann Ultraschalltechnik GmbH & Co. KG, Karlsbad-Ittersbach, Germany, with a generator frequency of 35 kHz and a maximum power of 1,000 W, equipped with a cylindrical ultrasonic horn with a diameter of 11 mm (Fig. 1(a) and (b)). The microstructured embossing tool was mounted on top of an additional support-plate, which also carries 2–3 alignment-pins (Fig. 1(a) and (c)).

Ultrasonic embossing system: (a) Schematic view of the complete system (b) Photograph of the ultrasonic horn during the embossing process (c) Photograph of the support plate with the embossing tool in the center and 3 alignment pins
To perform the ultrasonic embossing process, the multiwell plate was positioned upside down whereas the alignment-pins helped to lock the plate into position and to avoid any lateral movements while ultrasound was applied. After the positioning of the multiwell plate, the ultrasonic horn was pressed onto the backside of the multiwell plate by means of the integrated pneumatic cylinder and the ultrasonic converter was activated for typically less than 1 s. The oscillations of the converter were intensified by a mechanical booster and by the shape of the ultrasonic horn itself. The ultrasonic vibrations caused a local heating of the polymer and the softened bottom surface of one single well was pressed into the microstructured cavities of the embossing tool. Since the molten polymer has to be displaced by the embossing tool, the machine was operated in such a manner, that the oscillations were stopped as soon as the microstructured cavities of the mold are completely filled. To let the polymer cool down the ultrasonic horn remained in its position for another second and then the multiwell plate was removed to structure the next well. While the ultrasonic embossing process itself only lasts about 2 s, the removing and repositioning of the multiwell plate usually takes about 5–10 s. For a complete 96-well plate this adds up to about 15–20 min. However, this time might be reduced by an automated positioning system which also might improve the mechanical fixing of the multiwell plate to avoid lateral movements while the ultrasound is applied.
2.2.2 Microstructuring of the multiwell plates
The first experiments have been performed using an embossing tool (Fig. 2) which was an electroformed copy of the injection molding tool, which was used to produce the cell containers in our previous work (Giselbrecht et al. 2002; Gottwald et al. 2007). While the original tool was produced by micromilling techniques, the new embossing tool was manufactured by electroforming, i.e. filling the cavities of an already molded chip container with Ni (Wissmann et al. 2010). According to the dimensions of a single well of a standard 96-well cell culture plate, the embossing tool was machined to a diameter of 5 mm. The microstructures consisted of numerous square-shaped columns with an edge length of 300 μm and a height of 500 μm at a distance of 50 μm.

Electroformed embossing tool: (a) Top view of embossing tool of 5 mm diameter (b) Scanning electron micrograph of the embossing tool (c) Scanning electron micrograph of the microstructured multiwell plate
Since 500 μm deep microstructures may lead to a limited nutrient and gas supply of the cells inside the cavities due to a long diffusion path, the following experiments were performed using an embossing tool with a reduced height of 150 μm. This tool was fabricated from a stainless steel rod with a diameter of 5 mm by electric discharge machining using a cutting wire with a diameter of 100 μm, followed by electro polishing. To improve the removing process the design included a small draft angle. The resulting microstructures had an edge length of 300 μm and a distance of 130 μm (Fig. 3).

Embossing tool made by electric discharge machining: (a) Top view of the embossing tool of 5 mm diameter (b) Scanning electron micrograph of the microstructure (c) Scanning electron micrograph of the microstructured multiwell plate
For cell culture applications ultrasonic embossed multiwell cell culture plates (in the following: ULTECC-plates) were sterilized by γ-irradiation. Previous to cell seeding the air in the cavities of the microstructured 96-well plates was removed to ensure complete wetting of the microstructured surface area by applying a descending series of ethanol / water (distilled, sterile).
2.3 Cell culture conditions and induction of biotransformation enzymes
Human hepatoma cell line HepG2 was purchased from the American Type Culture Collection (LGC Promochem, Wesel, Germany). HepG2 cells were cultured in Minimum Essential Medium (MEM) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 1% (w/v) sodium pyruvate, 1% (w/v) non essential amino acids, 1% (w/v) glutamine and 10% (w/v) fetal bovine serum. The cells were maintained in a humidified 37°C incubator with 5% CO2, seeded at a density of 0.8 × 105 cells/cm² on cell culture flasks and split twice a week. Experiments were carried out with HepG2 cells from passage 9 to 15 in 3D culture configuration using microstructured ULTECC-plates and commercially available 96-well plates containing porous alginate sponges (AlgiMatrix™). Inoculation of the ULTECC-plates was accomplished by pipetting 200 μl cell suspension containing 105 cells into each well, while cell seeding in alginate sponges was performed according to the manufacturer’s protocol. In short, 105 cells in 30 μl culture medium were pipetted onto each alginate sponge and incubated for 10 min at 37°C in an incubator. Afterwards each well containing an inoculated sponge was filled up with 200 μl culture medium.
Based on the results of preliminary experiments with HepG2 cells (data not shown), induction of the investigated phase I and II enzymes was performed as described in the following: HepG2 cells were seeded into 96-well ULTECC-plates or alginate sponges as previously described and cultured for 24 h in an incubator. In order to induce biotransformation enzyme activity cells were treated with 2 μM 3-methylcholanthrene (3-MC) (1 mM stock solution in dimethyl sulfoxide diluted in culture medium) for another 24 h at 37°C. Thereafter, cell culture medium with 3-MC was removed and specific enzyme activity in HepG2 cells was determined as specified in section 2.5.2 and 2.5.3.
2.4 Confocal laser scanning microscopy
Examination of cell distribution and viability in the microstructured cavities of the 96-well plates was performed by simultaneous staining with the nuclear dyes Syto®16 (Molecular Probes®, Invitrogen, Karlsruhe, Germany) and propidium iodide (PI) as described in Altmann et al. (2008). In brief, cells were washed once with PBS and incubated with 5 μM Syto®16 and 1 μM PI in culture medium for 30 min at 37°C. For laser scanning microscopy the microstructured area was cut out of the plate after manufacturing, sterilized and inoculated with 105 HepG2 cells. Confocal laser scanning microscopy was carried out with a Leica TCS SP5 microscope.
2.5 Cell-based assays
2.5.1 alamarBlue® cell metabolism assay
Metabolic activity of HepG2 cells during 3D cell culture in ULTECC-plates and alginate sponges was assessed by the alamarBlue® assay. The alamarBlue® reagent comprises the redox-sensitive reporter dye resazurin, which is taken up by the cells and is reduced by mitochondrial respiration. The reduced product resorufin can be quantified in the supernatant by fluorimetry or colorimetry. For this purpose culture medium was replaced with medium containing 10% (w/v) alamarBlue® reagent (AbD Serotec, Düsseldorf, Germany). After incubation for 2 h at 37°C samples (100 μl) were transferred to microtiter plates and fluorescence was measured in a microplate reader (SPECTRA max M2, Molecular Devices) at λEx = 560 nm and λEm = 590 nm. Percentage of alamarBlue® reduction in the probes was calculated using a 100% reduced alamarBlue® control as reference.
2.5.2 Determination of enzyme activity of ethoxycoumarin-O-deethylase (ECOD) and ethoxyresorufin-O-deethylase (EROD)
ECOD and EROD are substrate specific cytochrome P450 (CYP) monooxygenases catalyzing reactions of the phase I metabolism. The deethylation of ethoxycoumarin by ECOD results in the formation of umbelliferone (7-hydroxycoumarin) conjugates, while deethylation of the substrate ethoxyresorufin is mediated by EROD and leads to the formation of resorufin conjugates. The activity of both enzymes in HepG2 cells was determined by fluorimetric quantification of the formed metabolites in the culture medium according to the protocol of Langsch and Bader (2001). For this purpose HepG2 cells were incubated with 10 μM 7-ethoxyresorufin or 50 μM 7-ethoxycoumarin at 37°C for 4 h. Since resorufin can also be metabolized by cytosolic diaphorase, 10 μM dicumarol was added to the incubation medium for the EROD assay to prevent further biotransformation of resorufin. After cell incubation with the respective substrates supernatant of each well was collected and stored at −20°C until analysis. In addition, cells were lysed for quantification of total protein content. For fluorimetric determination of the respective metabolites in the culture medium, samples were incubated over night at 37°C with 100 U/ml β-glucronidase (in 200 mM acetate buffer, pH 4.5) to cleave the umbelliferone and resorufin conjugates. Afterwards the samples were mixed with glycine buffer (1.6 M, pH 10.3), following determination of fluorescence at λEx = 360 nm and λEm = 460 nm for umbelliferone, and λEx = 530 nm and λEm = 580 nm for resorufin. Quantification of the metabolites was performed using an umbelliferone and resorufin standard curve. The determined metabolite concentrations were then normalized to incubation time with the respective substrates and total cellular protein content.
2.5.3 Determination of enzyme activity of UDP-glucuronosyltransferase (UGT) and sulfotransferase (SULT)
UGT and SULT are enzymes of the phase II metabolism and catalyze the glucuronidation of a substrate or the esterification with sulfate respectively. As both enzymes metabolize the substrate 4-methylumbelliferone (4-MU), the activity of UGT and SULT can be measured in one assay. In that case, UGT mediates the conjugation of 4-MU with UDP-glucuronic acid, which results in the formation of methylumbelliferone-7-glucuronide (MUG). SULT mediates the esterification of the substrate with sulfate leading to the formation of methylumbelliferone-7-sulfate (MUS). The substrate 4-MU, as well as the metabolites MUG and MUS were detected in culture medium by analytical HPLC. For this purpose, cells were incubated with 100 μM 4-MU (100 mM stock solution in dimethyl sulfoxide diluted in culture medium) for 24 h in an incubator at 37°C. After the incubation with 4-MU the medium was transferred into HPLC vials and cells were lysed for determination of total protein content. HPLC was performed on a Jasco HPLC system equipped with a binary high pressure mixer (dwell volume: 0.7 ml), an autosampler, a diode array UV detector and a Luna C18(2) column from Phenomenex (5 μ, 50 × 3 mm, with correspondent pre column, “Security Guard”). Eluent A: 90 vol% phosphate buffer, 10 vol% gradient grade acetonitrile. Eluent B: 60 vol% buffer, 40 vol% gradient grade acetonitrile. Phosphate buffer was prepared by dissolving 1.465 g NaH2PO4 and 1.869 g Na2HPO4 in 1 l water, adjusting pH to 7.0 and filtration. Eluent mixtures were degassed (vacuum and sonification) daily. Flow: 1 ml/min. Gradient: 0–0.3 min 100%A, 0.3–3.5 min increasing linearly to 100%B, 3.5–4.5 min holding 100%B, followed by column reequilibration. Injection volume: 25 μl. All components were detected at 320 nm. Retention times: Dead time: 0.4 min, MUG: 0.7 min, MUS: 2.2 min, and 4-MU: 3.0 min. Quantification was based on peaks heights. Calibration was performed daily using external standards of 50, 30, 20, and 10 μmol for MUS and MUG, and 100, 75, 50, 25 μmol for 4-MU in triplicates. Linear calibration curves passing through (0;0) were obtained with regression coefficients >0.999. The determined concentrations of MUG and MUS were normalized to total cellular protein content, and the results were expressed as fold-induction of vehicle-treated cells (negative control).
2.6 Protein determination
Cells were lysed with RIPA buffer consisting of 150 mM NaCl, 50 mM Tris–HCl, 0.25% (w/v) deoxycholic acid, 1 mM EDTA, 1% (w/v) NP40 and 0.1% (w/v) protease inhibitor cocktail, pH 7.4. Protein concentration in the cell lysates was determined with the Bio-Rad Protein Assay (Bio-Rad Laboratories, München, Germany), which is based on the method of Bradford. The assay was performed in microtiter plates according to the manufacturer’s protocol.
2.7 Statistical analysis
All experiments were performed in triplicates in three independent experiments unless otherwise indicated. Data are expressed as mean ± SEM. Data were compared for statistically significant differences using Mann–Whitney U-test (p < 0.05).
3 Results and discussion
3.1 Fabrication of microstructured multiwell plates
The experiments have been performed to investigate different types of 3D cell culture structures in multiwell plates and to test if ultrasonic embossing is an appropriate method to produce the desired microstructures. For that reason, the layout of the microstructures was not specially designed for the circular bottom of the wells. For the first experiments the embossing tool was machined to a diameter of 5 mm based on a rectangular molding tool. While the microstructures mainly consisted of square-shaped columns with an edge length of 300 μm, the border area of the microstructures comprised columns with unequal edge lengths or even a triangular shape.
Even though the side walls of the embossing tools with 500 μm height from the first experiments were very plane, it was very difficult to remove the 96-well plate after ultrasonic embossing due to the large total area of the side walls of about 85 mm². Embossing tool and microstructured multiwell plate are shown in Fig. 2.
Since 500 μm deep microstructures may lead to a limited nutrient and gas supply of the cells inside the cavities due to a long diffusion path, the following experiments were performed using an embossing tool with a reduced height of 150 μm. This tool (Fig. 3) was fabricated by electric discharge machining of a 5 mm in diameter stainless steel rod using a 100 μm cutting wire. Due to the reduced height and the increased thickness of the separation walls the total area of the side walls was about 17 mm². For further improvement of the removing process, the design also included a small draft angle and the surface of the tool was smoothened by electro polishing.
Due to the fact, that the polymer is molten locally, the size of the microstructures depends on the relation between the volume to be displaced by the solid columns of the embossing tool and the free volume in between, where the molten polymer can escape. For that reason, the ultrasonic embossing process was stopped as soon as all cavities of the molding tool were filled to avoid the formation of small beads of molten polymer along the edge of the molding tool. For this purpose, we used the build-in depth survey of the ultrasonic welding machine. In case of the embossing tool, which had a height of 150 μm, the penetration depth in relation to the original bottom of the multiwell was about 80 μm. However, in very few cases the separation walls between the individual microcavities have not been completely filled during the ultrasonic embossing process due to a different flow behavior of the melt. If there is a small tilt between the multiwell plate and the embossing tool, the ultrasonic vibrations will start at one side of the embossing tool resulting in an inhomogeneous temperature distribution of the melt.
In search of the lower limit for the size of the raised structures it has to be taken into account that the smaller the distance between the solid columns of the molding tool is, the larger is the cooling of the melt due to the larger relation between surface and volume of the melt. Schomburg et al. (2011) have produced structures down to 40 μm by using ultrasonic embossing; smaller structures could not be filled completely.
Our experiments have shown that for a good reproducibility of the embossing process it is very important to accurately lock the multiwell plate into position to avoid any lateral movements while the ultrasonic vibrations are applied. However the alignment pins used in our setup could not guarantee this in any case. Multiwell plates from different manufactures have small variations in diameter, draft and depth of the wells, which would require individual alignment pins for each manufacturer’s product. Additionally we found small deviations at different positions within one multiwell plate probably caused by anisotropic shrinkage during the manufacturing process of the standard multiwell plates. For that reason, an external support frame to lock the plate into position would be useful. The ultrasonic embossing also requires a plane backside of the multiwell plate to ensure a good coupling of the ultrasonic horn. However, some manufacturers use structured backsides on their multiwell plates.
3.2 Organization and viability of HepG2 cells in ULTECC-plates
Organization and viability of HepG2 cells in 3D culture were assessed by live/dead staining with the nuclear dyes Syto®16 (green fluorescence for living cells) and PI (red fluorescence for dead cells) as described in section 2.4. For optical analysis of adherent cells, the microstructured area of the ULTECC-plates was imaged with a laser scanning microscope. The resulting z-stacks are illustrated as horizontal and vertical maximum projections and shown in Fig. 4. The microscopic analysis revealed a uniform cell distribution within the microstructured area and the formation of adherent aggregates inside the microcavities after 48 h (Fig. 4(a) and (b)). Furthermore, HepG2 cells showed high viability after 48 h (Fig. 4(a)) and 4 days (Fig. 4(c)) of culture in ULTECC-plates with no detectable dead cells. The multicellular self-assembly in the microcavities of ULTECC-plates is consistent with findings of Lin et al. (2006), which described the hepatoma spheroid formation in hanging drops as a three-stage process, characterized by an initial rapid aggregation, a subsequent delayed period and a further tight compaction after 36 h of culture. Moreover, a compact aggregate formation in the microstructured plates was already observable after 24 h of culture (pictures not shown), while HepG2 cells in hanging drops formed only loose aggregates with individual cells identifiable after 24 h (Lin et al. 2006). The accelerated aggregate formation in our scaffolds might issue from the different culture methods.

Confocal images of the cell line HepG2 in microcavities of ULTECC-plates after 48 h ((a) and (b)) or 4 days ((c) and (d)) culture. Micrographs show maximum projections of green fluorescence (Syto®16, living cells) and red fluorescence (propidium iodide, dead cells, not detectable). (a) and (c) Top views. (b) and (d) Side views
3.3 alamarBlue® cell metabolic assay
Metabolic activity of HepG2 cells in ULTECC-plates was examined by the reductase-based alamarBlue® assay and compared to cultures in alginate sponges. As the alamarBlue® reagent is not cytotoxic, measurement of mitochondrial activity was repeated daily with the same cultures during a period of 4 days. At the end of the experiments cells were lysed and total protein content was quantified to compare protein content between different culture substrates at the last culture day.
Determination of alamarBlue® reduction in HepG2 cells revealed a higher cellular metabolic activity of the cells in ULTECC-plates compared to cultures in alginate sponges (Fig. 5). Furthermore, both culture configurations showed a continuous increase in alamarBlue® reduction indicating that cell proliferation occurred in both substrates during the whole culture period. Interestingly, total cellular protein content at day four of culture was similar in both cell culture substrates and averaged about 0.3 mg per well. Furthermore, the lower reduction rate of alamarBlue® in alginate sponges was accompanied by a distinct visible gradient of reduced alamarBlue® inside the sponge and non-reduced alamarBlue® in the supernatant. This stable diffusion gradient was detectable after 2 h of incubation when sampling was performed and even after 4 h incubation. As the alamarBlue® assay is based on the diffusion of the reporter dye resazurin into the cells and the subsequent release of the reduced form resorufin into the supernatant, this assay reflects the mass transport between cell aggregates and culture medium. The results of the alamarBlue® assay indicate that the lower reduction rate of the reporter dye in alginate sponges is rather due to an insufficient mixture of reduced and non-reduced alamarBlue® culture medium than on a lower proliferation rate of HepG2 cell aggregates in alginate sponges. Thus, it seems that despite the porous structure of the alginate sponge, mass exchange between sponge and culture medium was limited in the alginate sponges compared to ULTECC-plates.

alamarBlue® reduction by HepG2 cells cultured in ULTECC-plates and in alginate sponges
3.4 ECOD and EROD activity
Cytochrome P450 monooxygenases play an important role in the oxidative metabolism of a variety of exogenous and endogenous compounds. The resulting metabolites are directly excreted or undergo further conjugation with endogenous molecules in the phase II metabolism of biotransformation reactions. In the present work we examined the activities of the cytochrome P450 monooxygenases ECOD and EROD, which are mediated by Cyp1A1/2 and Cyp2B6 respectively. Both enzymes contribute to the drug metabolism, whereas especially Cyp1A2 is involved in the bioactivation of pro-carcinogens (Hewitt et al. 2007). Quantification of the metabolites resorufin and umbelliferone in the culture medium revealed a distinct higher activity of ECOD in HepG2 cells cultured in ULTECC-plates compared to cultures in alginate sponges (Fig. 6). In contrast, EROD activity showed no significant difference between both culture configurations, even though the amount of formed resorufin tended to be slightly higher in ULTECC-plates than in alginate sponges. Furthermore, HepG2 cells in microstructured plates showed a higher enzyme activity of ECOD compared to EROD after induction with 3-MC, whereas ECOD and EROD activities in alginate sponges were similar and lower than in ULTECC-plates.

ECOD and EROD activity in HepG2 cells cultured in microstructured 96-well plates and alginate sponges after 24 h induction with 3-MC. Since the metabolites resorufin and umbelliferone were not detectable in samples of untreated cells, the corresponding controls are not shown
The induction of both phase I enzymes in HepG2 cells by 3-MC has already been reported for monolayer cultures on gene expression level, even though fold-induction of CYB2B6 was significant lower than of CYP1A1/1A2 with the same inducer (Westerink and Schoonen 2007a). Three-dimensional culture of HepG2 cells revealed an inverse effect on protein level with a distinct higher ECOD activity compared to EROD, while cell aggregates in alginate sponges showed a lower and comparable activity of both enzymes. The higher ECOD activity in ULTECC-plates may be due to a limited diffusion of the inducer or substrate through the alginate sponge into the cell aggregate and/or a slower diffusion/release of the formed metabolite into the supernatant during incubation time, which was 4 h. Such a restricted mass exchange was already observable in the alamarBlue® assay (section 3.3) and might explain the different results. To clarify the underlying mechanism that leads to the high ECOD activity of HepG2 cells in ULTECC-plates, more studies concerning gene expression level of Cyp2B6 in these cells have to be done.
3.5 UGT and SULT induction
Glucuronidation is one of the major detoxification systems in humans and is mediated by UDP-glucuronosyltransferases. The conjugation of various xenobiotics and endogenous compounds with UDP-glucuronic acid is mediated by UDP-glucuronosyltransferases and results in a higher polarity, as well as water solubility of the substrates, which leads to a facilitated excretion of the substance (Burchell and Coughtrie 1989; McCarver and Hines 2002). Sulfotransferases on the other hand catalyze the sulfate conjugation of a variety of endobiotics and exogenous chemicals using 3′-phosphoadenosine-5′-phosphosulfate as donor. Reactions of these phase II drug-metabolizing enzymes are not only associated with detoxification and pharmacological inactivation, but also with the bioactivation of xenobiotics resulting in reactive metabolites (Glatt 2000; McCarver and Hines 2002). The activity of the enzymes UGT and SULT in HepG2 cells was determined by quantification of the formed metabolites MUG and MUS in the culture medium after induction with the model inducer 3-MC. The results revealed a higher UGT induction in HepG2 cells cultured in ULTECC-plates compared to cells in alginate sponges (Fig. 7). Moreover, treatment with 3-MC resulted in a 1.9-fold higher UGT activity in HepG2 cells, whereas UGT induction in alginate cultures was 1.3-fold of untreated controls. As already discussed in section 3.3 and 3.4, it can not be excluded that the higher UGT activity in ULTECC-plates may be a result of different mass exchange conditions between cell aggregates and culture medium in both substrates, causing different absorption or release rates of the inducer or metabolite. The results are consistent with other studies showing the induction of the isoforms UGT1A1 and UGT1A6 in HepG2 monolayer cultures with 3-MC on mRNA level (Westerink and Schoonen 2007b). Moreover, previous work revealed an upregulation of the constitutive gene expression of UGT1A1 in HepG2 cells cultured in 3D microstructured polymer chips compared to conventional monolayer cultures (Altmann et al. 2008). If the higher induction of the overall UGT activity in ULTECC-plates compared to alginate sponges is accompanied by a higher gene expression or is a result of a higher activity of the corresponding enzyme, has to be evaluated in further studies comparing mRNA expression levels in both culture configurations. In contrast, SULT induction in HepG2 cells by 3-MC did not occur in any of the culture configurations. Another inducer namely β-naphtophflavone, which was reported to increase SULT1A1 and SULT1A2 transcript levels in HepG2 (Westerink and Schoonen 2007b), did not show any effect on overall SULT activity in this cells in the present work (data not shown). However, data of other groups were generated with monolayer cultures and are difficult to compare with three-dimensional culture configurations, as microenvironment concerning substrate or metabolite concentration, pH and oxygen concentration can vary between different culture configurations. In this context, Hewitt and Hewitt (2004) could show that the mRNA expression levels and activities of biotransformation enzymes in HepG2 cells depend not only on the cell source, but also on the culture conditions.

UGT and SULT induction in HepG2 cells cultured in microstructured 96-well plates and alginate sponges after treatment with 3-MC. Results are presented as fold induction of untreated controls (untreated controls were defined as 1×, marked as dotted line)
4 Conclusions
Since the introduction of three-dimensional cell culture techniques for the generation of multicellular aggregates, a vast number of studies using different types of adult, progenitor and stem cells showed that this culture configuration represents a better in vitro tissue model compared to conventional monolayer cultures. In the present work, we describe for the first time a method for the fabrication of 3D cell culture substrates in a multiwell plate format for cell-based HTS applications by microstructuring commercially available 96-well cell culture plates using ultrasonic embossing. The described method comprises a local heating of the polymer by ultrasonic vibrations and the subsequent microstructuring of the locally softened bottom surface of a single well by a special designed embossing tool. This might be a cost effective method for prototype or small scale fabrication, in particular since a change of the design of the microstructures only affords the replacement of the single embossing tool.
The geometry of the microstructured area comprising cubic microcavities was based on the design of a culture system developed in the Karlsruhe Institute of Technology, which proved to be suitable for the generation of multicellular aggregates (Altmann et al. 2008; Gottwald et al. 2007; Rieke et al. 2008). As former work with the human liver-derived cell line HepG2 in 3D cell culture configuration revealed an increased expression of liver-specific genes compared to monolayer culture (Altmann et al. 2008; Horiuchi et al. 2009), we used HepG2 cells as in vitro model to study the hepatocellular metabolic functions in our microstructured 3D substrates and to examine the suitability of the culture system for screening purposes. In addition, we compared our system with a commercially available 3D culture system based on porous alginate sponges in 96-well plates.
Our results showed a uniform cell distribution within the microstructured area of the modified multiwell plates and the formation of adherent cell aggregates exhibiting high viability inside the microcavities. Moreover, the performed cell-based assays revealed a higher metabolic activity of HepG2 cells in our culture system compared to alginate sponges. As these metabolism assays were based on direct incubation of the cells with specific substrates and the subsequent quantification of the formed metabolites in the supernatant, our findings indicate that the higher cell metabolism was probably due to a better mass transport between microcavities and culture medium as compared to porous alginate sponges. A similar situation was reported for the drug clearance in liver slices in comparison to isolated cells, which showed a lower metabolism rate than the isolated hepatocytes and may result from a slower mass transport between several cell layers (Carlile et al. 1999). Thus, the microenvironment of 3D cell culture configurations is an important parameter which influences the accessibility of compounds and the release of the formed metabolites inside the cell aggregates. This is especially important in terms of the interpretation of data obtained from cell-based metabolism studies with highly metabolic active cells like hepatocytes. Compared to commercially available 3D cell culture systems, which mainly comprise sponges, hydrogels and nanofibers, mass exchange in our modified plates was only limited by the mass transport within the cell aggregate inside the microcavities and not by the diffusion through a gel or fiber matrix.
Since we modified standard polystyrene cell culture plates, our system might be useful for all kinds of cell cultures which need a 3D culture configuration to induce, restore or maintain the differentiated status. Furthermore, many standardized techniques for cell analysis used so far in conventional monolayer culture, like cell lysis for mRNA or protein extraction, quantification of secreted proteins into the culture medium, are often difficult to transfer to 3D culture systems, especially in gel-based systems as gels often hinder the accessibility of the cells. We therefore think that the presented 3D cell culture substrates represent a helpful tool not only in drug discovery and toxicological studies, but also in the research fields of cancer and stem cell research, as well as tissue engineering.
References
S.F. Abu-Absi, J.R. Friend, L.K. Hansen, W.S. Hu, Exp. Cell Res. 274, 56 (2002)
B. Altmann, S. Giselbrecht, K.F. Weibezahn, A. Welle, E. Gottwald, Biomed. Mater. 3, 034120 (2008)
B. Burchell, M.W. Coughtrie, Pharmacol. Ther. 43, 261 (1989)
D.J. Carlile, N. Hakooz, J.B. Houston, Drug Metabol. Dispos. 27, 526 (1999)
K. Cheng, Y. Lai, W.S. Kisaalita, Biomaterials 29, 2802 (2008)
S. Giselbrecht, E. Gottwald, G. Knedlitschek, K. F. Weibezahn, A. Welle, A. E. Guber, D. Herrmann, A. Muslija, W. K. Schomburg, Biomed. Tech. (Berl) 47 Suppl 1 Pt 1, 373 (2002)
H. Glatt, Chem. Biol. Interact 129, 141 (2000)
E. Gottwald, S. Giselbrecht, C. Augspurger, B. Lahni, N. Dambrowsky, R. Truckenmuller, V. Piotter, T. Gietzelt, O. Wendt, W. Pfleging, A. Welle, A. Rolletschek, A.M. Wobus, K.F. Weibezahn, Lab Chip 7, 777 (2007)
N.J. Hewitt, P. Hewitt, Xenobiotica 34, 243 (2004)
N.J. Hewitt, R. de Kanter, E. LeCluyse, Chem. Biol. Interact. 168, 51 (2007)
S. Horiuchi, S. Ishida, T. Hongo, Y. Ishikawa, A. Miyajima, J. Sawada, Y. Ohno, K. Nakazawa, S. Ozawa, Biochem. Biophys. Res. Commun. 378, 558 (2009)
B.A. Justice, N.A. Badr, R.A. Felder, Drug Discov. Today 14, 102 (2009)
A. Khademhosseini, R. Langer, J. Borenstein, J.P. Vacanti, Proc. Natl. Acad. Sci. U. S. A. 103, 2480 (2006)
P. Khuntontong, T. Blaser, W.K. Schomburg, IEEE Trans. Electron. Packag. Manuf. 32, 152 (2009)
N. Koide, T. Shinji, T. Tanabe, K. Asano, M. Kawaguchi, K. Sakaguchi, Y. Koide, M. Mori, T. Tsuji, Biochem. Biophys. Res. Commun. 161, 385 (1989)
L.A. Kunz-Schughart, J.P. Freyer, F. Hofstaedter, R. Ebner, J. Biomol. Screen. 9, 273 (2004)
J.L. Lam, L.Z. Benet, Drug Metabol. Dispos. 32, 1311 (2004)
A. Langsch, A. Bader, Biotechnol. Bioeng. 76, 115 (2001)
J. Lee, M.J. Cuddihy, N.A. Kotov, Tissue Eng. Part B Rev. 14, 61 (2008)
R.Z. Lin, L.F. Chou, C.C. Chien, H.Y. Chang, Cell Tissue Res. 324, 411 (2006)
S.-J. Liu, Y.-T. Dung, Polym. Eng. Sci. 45, 915 (2005)
D.G. McCarver, R.N. Hines, J. Pharmacol. Exp. Ther. 300, 361 (2002)
H. Mekaru, O. Nakamura, O. Maruyama, R. Maeda, T. Hatt, Microsyst. Technol. 13, 385 (2007)
J.K. Park, D.H. Lee, J. Biosci. Bioeng. 99, 311 (2005)
M. Rieke, E. Gottwald, K.F. Weibezahn, P.G. Layer, Lab Chip 8, 2206 (2008)
W.K. Schomburg, K. Burlage, C. Gerhardy, Micromachines 2, 157–166 (2011). doi:3390/mi2020157
M.G. Soars, D.F. McGinnity, K. Grime, R.J. Riley, Chem. Biol. Interact 168, 2 (2007)
R. Truckenmüller, Y. Cheng, R. Ahrens, H. Bahrs, G. Fischer, J. Lehmann, Microsyst. Technol. 12, 1027 (2006)
M. Vermeir, P. Annaert, R.N. Mamidi, D. Roymans, W. Meuldermans, G. Mannens, Expert Opin. Drug Metab. Toxicol. 1, 75 (2005)
K.F. Weibezahn, G. Knedlitschek, H. Dertinger, K. Schubert, W. Bier, T. Schaller. International patent WO 93/07258, Cell culture substrate (1993)
W.M. Westerink, W.G. Schoonen, Toxicol. Vitro 21, 1581 (2007a)
W.M. Westerink, W.G. Schoonen, Toxicol. Vitro 21, 1592 (2007b)
M. Wissmann, M. Guttmann, M. Hartmann, A. Hofmann and B.Hummel; Courtois, B. [Publ.] Collection of papers presented at the Symp. On design, test, integration and packaging of MEMS/MOEMS (DTIP 2010), Sevilla, E, May 5–7, 2010 Grenoble: EDA Publ. (2010) 278–83; ISBN 978-2-35500-011-9
M. Worgull, Hot embossing: Theory and technology of microreplication (William Andrew, Oxford, 2009)
F.J. Wu, J.R. Friend, R.P. Remmel, F.B. Cerra, W.S. Hu, Cell Transplant 8, 233 (1999)
Acknowledgement
The authors thank Simone Weigel for performing HPLC, Anja Becker for technical assistance, Daniel Münch for the ultrasonic embossing of numerous cell culture plates and Gregor Schlingloff for helpful discussions.
This work was supported by the BMBF (grant no. 03ZIK-465).
Author information
Authors and Affiliations
Corresponding author
Additional information
Brigitte Altmann and Ralf Ahrens contributed equally to this work.
Rights and permissions
About this article
Cite this article
Altmann, B., Ahrens, R., Welle, A. et al. Microstructuring of multiwell plates for three-dimensional cell culture applications by ultrasonic embossing. Biomed Microdevices 14, 291–301 (2012). https://doi.org/10.1007/s10544-011-9605-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10544-011-9605-8