
Abstract
High Selenium Yeast (SeY) serves many important roles with respect to the maintenance of normal nervous system functioning. Studies have reported the nerve inflammation induced by Aluminum (Al) was associated with the increase of mortality. However, in-depth studies are required to verify the hypothesized neuro-protective efficacy of SeY against Al-induced cerebral damage through modulation of the inflammatory response. Here, mice were treated with SeY (0.1 mg/kg) and/or Al (10 mg/kg) by oral gavage for 28 days. Inflammation was assessed by histopathological examination and expression of biomarkers for inflammation. Furthermore, the oxidation–reduction levels and the NO production were assessed using diagnostic kits and RT-PCR. The data indicated that SeY significantly protected cerebrum against Al-induced pathological changes, in addition to the disordered expression of biomarkers of inflammation, the imbalance of oxidation–reduction, and the increase of NO production. Therefore, the chemoprotective potential of SeY against Al-induced cerebral inflammation via restore the levels of oxidation–reduction and the generation of NO was demonstrated.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In the past decades, worldwide production of aluminum (Al) has rapidly increased, from 5 million tons in 1960 to 425 million tons in 2002, with developing countries increasing their domestic consumption by more than 350% (Ingerman et al. 2008). As a result of the increased production and use of Al, increasing evidence of toxic exposures to animals has been observed, likely through the ingestion of contaminated food and water. These exposures have been implicated in the emergence of several biochemical and metabolic pathologies. Several studies have described pro-oxidant effects (Ruipérez et al. 2012), modifications of the essential metal homeostasis (Walton 2012), double strand DNA breaks (Sappino et al. 2012), and altered release of some cytokines involved in major inflammatory pathways (Darbre et al. 2013; Mannello et al. 2013). Oxidative damage to cells in the murine hippocampus induced by Al has been clearly demonstrated (Ding and Yang 2010). It has been reported Al inhibited rat osteoblastic differentiation through inactivation of the Wnt/β-catenin signaling pathway (Sun et al. 2017). The PGD2-DP1 signaling pathway protects against the neuronal injury to primary rat hippocampal neurons in vitro resulting from exposure to high levels of Al (Ma et al. 2016). However, the effects of Al-induced inflammation in the cerebrum are currently not well understood.
Selenium (Se) is believed to support various important cellular and organismal functions as an essential trace element in many organisms, including avian and mammalian species (Kryukov et al. 2003). With a number of studies demonstrating its importance to normal physiological functioning of the brain, Se appears to have serve in a multifaceted role in the nervous system. The antioxidant activities of Se in the CNS are well established, and lower levels of Se have been associated with brain injury (Duntas and Benvenga 2015). Studies have reported therapeutic benefits after high doses of Se were administered to patients with inflammatory processes (Angstwurm et al. 2007; Huang et al. 2013). Deficiencies of Se can result in increased inflammation. Mertens et al. reported that reduced concentrations of Se were correlated with increased oxidative stress and biomarkers of inflammation. Oxidative stress resulting from suboptimal Se levels might contribute to the damage of important proteins (Mertens et al. 2015). Cao et al. reported that Se deficiency induced inflammatory responses in broiler chickens (Cao et al. 2017). Deficiencies in Se are believed to increase the levels of inflammatory cytokines including COX-2, iNOS, IL-1β and IL-6 (Zhu et al. 2015). It has also been demonstrated that Se can alleviate Pb-induced increases of inflammation factors (TNF-a, NF-kB, COX-2, iNOS) in chickens (Wang et al. 2015). Protective roles of Se on nitric oxide (NO) and gene expression of inflammatory cytokines induced by Cd exposure has been demonstrated by Liu et al. (Shuang et al. 2015). Furthermore, Se can antagonize the inflammatory response and oxidative stress induced by Hg exposure (Ansar 2016). High levels of dietary Se intake has been associated with a beneficial body composition profile (Wang et al. 2016). Adequate Se dietary supplementation can increase the anti-oxidative levels and enhance an anti-inflammatory state (Huang et al. 2012). Selenium-yeast (SeY), the organic from of Se, has many advantages over sodium selenite (Na2SeO3) and sodium selenate (Na2SeO4). Organic Se from SeY can be stored in a protein pool that increases its bioavailability (Rajashree et al. 2014). Animal studies have demonstrated that the bioavailability of organic forms of Se is higher than that of inorganic forms (Edens 1995). However, the protective efficacy of SeY to the cerebrum from damage caused by toxic exposure to Al has remained elusive.
Although numerous studies have corroborated the hypothesis that Al-induced inflammation is likely the cause of the associated neurotoxicity, studies examining the antagonism of the cerebral toxicity by SeY are few and far between. The aim the present study was to explore the effects of SeY supplementation on the molecular mechanism of Al induced cerebral inflammation.
MM
Compliance with ethical standards
All animal studies reported here were approved by the Institutional Animal Care and Use Committee of the Foshan University.
Animals and experimental design
A total of 40 four-week-old male Kunming mice (18–23 g) were allowed a 7 day period for acclimation to their housing prior to initiation of the study. The animals were housed at constant temperature of 22 ± 2 °C, with a relative humidity of 50 ± 15%, and a 12-h light/dark cycle. Beginning at day 0, body weights were measured daily, and the consumption of drinking water and feed (provided ad libitum) were measured daily. Mice were individually housed in polycarbonate cages. Prior to initiation of the experiment, baseline levels of SeY and Al were measured. The results showed the contents of SeY and Al were 0.102 and 0 mg/kg, respectively.
Mice were divided into four groups as follows:
-
Group 1 (C) autoclaved water twice daily for 28 days.
-
Group 2 (SeY) autoclaved water with 0.1 mg/kg SeY for 28 days.
-
Group 3 (Al) autoclaved water with 10 mg/kg of Al for 28 days.
-
Group 4 (SeY + Al) autoclaved water with 10 mg/kg of Al and 0.1 mg/kg SeY for 28 days.
At the end of the experiment, mice were fasted before the day of sacrifice, and cerebrums were carefully dissected out, hold in − 80 °C after weighing.
Determination of total protein
Concentrations of total protein from tissues were measured using protein assay kits, and BSA was used to construct the standard curve.
Measurement of redox levels
Levels of H2O2, SOD, CAT, and t-AOC activities were measured using diagnostic kits provided by the Jiancheng Biotechnology Research Institute. Measurements were performed according to the manufacturer’s protocol.
Determination of cardiac NO concentrations and NOS activities
The concentrations of NO and activity levels of NOS were assessed using a commercially available kit from the Jiancheng Bioengineering Institute. Tissues were homogenized according to the manufacturer’s instructions. The NOS Activity Detection kit measures total activity of iNOS and tNOS (total NOS). The cNOS (constitutive NOS) activity was calculated by subtracting iNOS from tNOS.
qRT-PCR analysis
Total RNA was extracted from tissues, and the complementary DNA was synthesized using a RevertAid first strand cDNA synthesis kit. Gene expression levels were measured by qRT-PCR using a Light Cycler® 480 System and the Fast Universal SYBR Green Master Mix. Primer Analysis Software was used to design target specific oligonucleotide primers. The primers were commercially synthesized by the Beijing Genomics Institute Co., Ltd., China. A housekeeping gene, GAPDH, was used as an internal reference. The relative abundances of mRNA for each gene was calculated using the 2−ΔΔCt method, accounting for gene specific efficiency, and was normalized to the mean expression of GAPDH.
Statistical analysis
GraphPad Prism 7.0 (GraphPad Software Inc., USA) and SPSS 20 were used to test the effects of the dietary SeY and Al levels on measures. Multiple mean comparisons were performed using One-way ANOVA. Data are presented as the mean ± S.D. and values were considered statistically significant if P < 0.05.
Results
Histopathological analyses of cerebral tissues
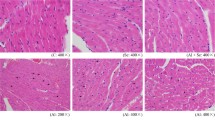
Histological analysis of cerebrum sections obtained from the control and SeY groups resulted in normal histological structure after 28 days of treatment (Fig. 1c, d). Following exposure to a toxic dose of Al, the cerebrum had significant abnormalities, including infiltration of inflammatory cells, neuronal shrinkage, and morphological changes associated with pyknosis. The neurons exhibiting atrophy were surrounded by numerous glial cells (Fig. 1a). In the SeY + Al treated group, limited neuronal shrinkage was observed, and the histopathological scores were improved over that of the Al treated group (Fig. 1b). These results indicated that histopathological changes in the cerebrum induced by Al, particularly inflammation, could be ameliorated by dietary SeY supplementation.

Effects of SeY and/or Al on the histopathology in the murine cerebrum (× 400). a Al group, the arrows are neurons and surrounded by numerous glial cells; b SeY+ Al group, the arrows pointed at limited neuronal shrinkages; c C group; d SeY group
Detection of inflammatory mediators in cerebral tissues
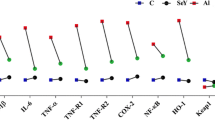
To characterize the inflammatory response, levels of inflammation cytokines (IL-1β, IL-6, TNF-α, TNF-R1, TNF-R2, COX-2, NF-κB, HO-1, Keap1 and Nrf2) were examined in the experimental mice. As is presented in Fig. 2, in comparison with the C group, 10 mg/kg Al significantly up-regulated the expression of IL-1β (217%), IL-6 (516%), TNF-α (167%), TNF-R1 (205%), TNF-R2 (158%), COX-2 (144%), NF-κB (311%), HO-1 (139%) and Nrf2 (136%) mRNA. However, this up-regulation was suppressed by SeY supplementation. The mRNA expression level of keap1 was significantly decreased (P < 0.05) to 67.8% of that in the Al treated animals compared with the C group. No significant effect on the mRNA expression of inflammatory cytokines was observed in the SeY alone group. The results indicated that the effects of Al on cerebral inflammatory cytokines can be reversed by dietary supplementation with SeY.

Effects of SeY and/or Al on the mRNA expression of inflammatory cytokines
Detection of antioxidant-related factors in cerebrum
To assess the degree of oxidative stress in Al and/or SeY treated mice, levels of H2O2, as well as t-AOC, CAT and SOD activities were examined. As is presented in Fig. 3, relative to the C group, the levels of H2O2 were significantly increased (P < 0.05) by 237% when treated with the Al diet. When SeY was added to the diet, H2O2 levels returned to normal. The activities of CAT, t-AOC and SOD were significantly down-regulated to 64.6, 59.3 and 71.0% (P < 0.05) in Al treated animals. Interestingly, dietary supplementation with SeY alone did not result in a measurable effect. This suggests that the redox status may be regulated by the SeY pretreatment, thus attenuating the toxic effects of Al on the antioxidant index.

Effects of SeY and/or Al on the generation of NO
Detection of NO production in cerebrum
The levels of NO in cerebrum when the mice were provided with a SeY and/or Al containing diet were measured The concentrations of NO and (i,t,c)-NOS activities in cerebrum are presented in Fig. 4. Compare with the C group, the concentration of NO was significantly up-regulated to 144% when treated with Al, while SeY supplementation decreased the content of NO to a basal level.

Effects of SeY and/or Al on markers of oxidation–reduction status
To further determine whether the suppression of NO production is due to the mRNA expression of NOS and NOS activities, (i,n,c)-NOS gene expression was assayed. It was observed that, compared with the C group, the mRNA expression of (i,n,c)-NOS were significantly up-regulated to 658, 161 and 290% (P < 0.05), respectively, in mice provided the Al containing diet. The activities of (i,t,c)-NOS were up-regulated to 157, 122 and 202%, respectively. Furthermore, the mRNA expression of (i,n,c)-NOS and the activities of (i,t,c)-NOS were not significantly different from the C group. These results suggest that treatment with SeY can restore NOS activities and NOS mRNA expressions altered by Al toxicity.
Discussion
Here, 3 unique metabolic responses in mice were demonstrated in response to the addition of dietary SeY and/or Al. The data showed that SeY can reduce the magnitude of the inflammatory reaction induced by Al through the modulation of the gene expression of 10 inflammatory factors. Furthermore, SeY was demonstrated to antagonize the oxidative stress caused by Al supplementation by improving the content/activity of 4 antioxidant enzymes, including t-AOC, CAT, H2O2 and SOD in the murine cerebrum. It was also demonstrated that SeY supplementation is capable of down-regulating the activity and content of NOS to resist the increase of NO production caused by Al. Therefore, the data presented here indicated that dietary supplementation with SeY relieved the inflammatory response through the regulation of oxidative stress and production of NO, therefore reducing the degree of tissue damage caused by Al.
Neuro-inflammation has been implicated as a primary mechanism leading to Al-induced neurotoxicity (Zheng et al. 2016). The increased expression of TNF-α likely plays a crucial role in the inflammation-mediated neurodegeneration associated with Al neurotoxicity (Johnson and Sharma 2003). Furthermore, IL-1β, NF-κB and COX-2 were significantly up-regulated when brain tissues were exposed to Al (Lukiw et al. 2005). In the present experiment, similar to what others have previously reported, our data showed that Al caused the dysregulation of pro-inflammatory (TNF-α, IL-6, COX-2, IL-1β) cytokines. Histopathologic examination revealed a large number of glial cells surrounding damaged neurons following exposure to Al alone. Therefore, it was hypothesized that Al induced inflammatory responses further induced cerebral injury. These data indicated the supplementary of SeY could inhibit the Al-induced inflammatory response via modulation the inflammatory mediators. There is an inverse relationship observed between Se levels and the occurrence of cerebral damage. Several other studies have suggested anti-inflammatory mechanisms of action that are considered to be related to the suppression of pro-inflammatory cytokine production, such as TNF-α, IL-6, NF-κB, IL-1β and COX-2 (Huang et al. 2012; Li et al. 2017b). Similar to previous reports, increases of pro-inflammatory factors, including TNF-α, IL-6, NF-κB, IL-1β and COX-2 were significantly down-regulated by SeY treatment.
A remarkable increase in lipid peroxidation was observed following Al exposure, which resulted in neuronal damage (Li et al. 2017a). The overload of Al can decrease SOD activity and increase MDA content in the rat nervous system (Pan et al. 2015). A significant depletion of CAT and GSH in brain caused by Al exposure resulted in further neurotoxicity (Li et al. 2017a). In addition, previous studies have also demonstrated the anti-oxidative mechanisms of Se (Xiang et al. 2017; Zhang et al. 2017). Here, oxidative stress was detected in cerebral tissues from mice treated with Al, which was reflected by reductions in CAT, T-AOC and SOD contents, as well as in rising levels of H2O2. Interestingly, the oxidative reductase targets returned to normal levels following SeY supplementation, which was observed by others as well. These findings suggested that dietary intake of SeY may have protective effects against the cerebral oxidative stress caused by Al.
Improvements of NO bioavailability have been identified as a key point at nerve tissue actions. An important intercellular messenger, NO is known to be widely involved in neurophysiological functions. Nitric oxide is the smallest signaling molecule within the central nervous system, and is synthesized under the action of the NO synthase (NOS) enzyme. There are three NOS isoforms: neuronal (nNOS), inducible (iNOS), and endothelial (eNOS) (Hosny et al. 2018). Typically, only cNOS is expressed in tissue. When the body is stimulated by inflammation for example, iNOS is produced, which further induces the transcription of iNOS, in turn catalyzing the production of large amounts of NO. A previous study by Macmicking et al. demonstrated that when LPS stimulation is used to induce inflammation, reduced mortality was observed in iNOS knockout mice (JD et al. 1995). It was reported that increasing concentrations of NO synthesis and oxidative stress were correlated with each another in children (Codoñer-Franch et al. 2011). The additional production of NO contributes to neuronal damage in astrocytes (Colombo et al. 2012). This is also consistent with the increase in glial cells observed here in the Al treatment group.
In order to investigate a possible correlation between the increase in NO production and the up-regulation of NOS activity and subtype gene expression, the effect of Al exposure on NOS activity and its subtype mRNA expression levels in mouse cerebrum was examined in Al exposed mice. Following Al exposure, the production of NO increased, which coincided with the increase in (i.t.c)-NOS gene expression and activities. These results were similar to previously published research examining whether excessive NO induced tissue inflammation in mice (Li et al. 2017c). The results indicated that the metabolic disturbance of the NO system is a leading cause of Al-induced inflammatory responses. Furthermore, it has been reported that Se can regulate the activity of NOS and the generation of NO, thus regulating the balance of oxidation–reduction in vivo (Chen et al. 2014; Zhao et al. 2014). In the present study, dietary supplementation with SeY restored the increase in NO production, the activities of (i,t,c)-NOS, and gene expression of (i,t,c)-NOS, which indicated that supplementary SeY could inhibit the Al-induced inflammatory responses via the modulation of NO production.
In summary, Al exposure induced cerebral inflammation through alterations in the balance of oxidation–reduction, and the enhancement of NO production. Alternatively, SeY significantly reduced the Al-induced damage. This study provides novel insights into the cerebral inflammatory pathology caused by Al exposure, and highlighted a potential protective role for dietary supplementation with SeY.
References
Angstwurm MW et al (2007) Selenium in Intensive Care (SIC): results of a prospective randomized, placebo-controlled, multiple-center study in patients with severe systemic inflammatory response syndrome, sepsis, and septic shock. Crit Care Med 35:118
Ansar S (2016) Effect of selenium on the levels of cytokines and trace elements in toxin-mediated oxidative stress in male rats. Biol Trace Elem Res 169:129–133
Cao C et al (2017) Inflammatory response occurs in veins of broiler chickens treated with a selenium deficiency diet. Biol Trace Elem Res 173:1–9
Chen X, Yao H, Yao L, Zhao J, Luan Y, Zhang Z, Xu S (2014) Selenium deficiency influences the gene expressions of heat shock proteins and nitric oxide levels in neutrophils of broilers. Biol Trace Elem Res 161:334–340
Codoñer-Franch P, Tavárez-Alonso S, Murria-Estal R, Megías-Vericat J, Tortajada-Girbés M, Alonso-Iglesias E (2011) Nitric oxide production is increased in severely obese children and related to markers of oxidative stress and inflammation. Atherosclerosis 215:475–480
Colombo E et al (2012) Stimulation of the neurotrophin receptor TrkB on astrocytes drives nitric oxide production and neurodegeneration. J Exp Med 209:521
Darbre PD, Mannello F, Exley C (2013) Aluminium and breast cancer: sources of exposure, tissue measurements and mechanisms of toxicological actions on breast biology. J Inorg Biochem 128:257–261
Ding R, Yang Y (2010) Aluminum chloride induced oxidative damage on cells derived from hippocampus and cortex of ICR mice. Brain Res 1324:96–102
Duntas LH, Benvenga S (2015) Selenium: an element for life. Endocrine 48:756–775
Edens FW (1995) Practical applications for selenomethionine: broiler breeder reproduction. In: Proceeding of the 18th annual symposium: nutritional biotechnology in the feed and food industry. Nottingham University Press, Nottingham, pp 29–42
Hosny EN, Sawie HG, Elhadidy ME, Khadrawy YA (2018) Evaluation of antioxidant and anti-inflammatory efficacy of caffeine in rat model of neurotoxicity. Nutr Neurosci. https://doi.org/10.1080/1028415x.2018.1446812
Huang Z, Rose AH, Hoffmann PR (2012) The role of selenium in inflammation and immunity: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 16:705–743
Huang TS, Shyu YC, Chen HY, Lin LM, Lo CY, Yuan SS, Chen PJ (2013) Effect of parenteral selenium supplementation in critically ill patients: a systematic review and meta-analysis. PLoS ONE 8:e54431
Ingerman L, Jones DG, Keith S, Chappell L, Rosemond ZA (2008) Toxicological profile for aluminum. ATSDR, Division of Toxicology and Environmental Medicine, Atlanta
Jd M et al (1995) Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell 81:641–650
Johnson VJ, Sharma RP (2003) Aluminum disrupts the pro-inflammatory cytokine/neurotrophin balance in primary brain rotation-mediated aggregate cultures: possible role in neurodegeneration. Neurotoxicology 24:261–268
Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigó R, Gladyshev VN (2003) Characterization of mammalian selenoproteomes. Science 300:1439–1443
Li L et al (2017a) Phenolic alkaloid oleracein E attenuates oxidative stress and neurotoxicity in AlCl3-treated mice. Life Sci 191:211–218
Li X et al (2017b) Effects of selenium–lead interaction on the gene expression of inflammatory factors and selenoproteins in chicken neutrophils ☆. Ecotoxicol Environ Saf 139:447–453
Li XN, Lin J, Xia J, Qin L, Zhu SY, Li JL (2017c) Lycopene mitigates atrazine-induced cardiac inflammation via blocking the NF-κB pathway and NO production. J Funct Foods 29:208–216
Lukiw WJ, Percy ME, Kruck TP (2005) Nanomolar aluminum induces pro-inflammatory and pro-apoptotic gene expression in human brain cells in primary culture. J Inorg Biochem 99:1895–1898
Ma J et al (2016) Effect of the PGD2-DP signaling pathway on primary cultured rat hippocampal neuron injury caused by aluminum overload. Sci Rep 6:24646
Mannello F, Ligi D, Canale M (2013) Aluminium, carbonyls and cytokines in human nipple aspirate fluids: possible relationship between inflammation, oxidative stress and breast cancer microenvironment. J Inorg Biochem 128:250–256
Mertens K et al (2015) Low zinc and selenium concentrations in sepsis are associated with oxidative damage and inflammation. Br J Anaesth 114:990–999
Pan Y et al (2015) Beraprost sodium protects against chronic brain injury in aluminum-overload rats. Behav Brain Funct 11:1–8
Rajashree K, Muthukumar T, Karthikeyan N (2014) Comparative study of the effects of organic selenium on hen performance and productivity of broiler breeders. Br Poult Sci 55:367–374
Ruipérez F, Mujika JI, Ugalde JM, Exley C, Lopez X (2012) Pro-oxidant activity of aluminum: promoting the Fenton reaction by reducing Fe(III) to Fe(II). J Inorg Biochem 117:118–123
Sappino AP, Buser R, Lesne L, Gimelli S, Béna F, Belin D, Mandriota SJ (2012) Aluminium chloride promotes anchorage-independent growth in human mammary epithelial cells. J Appl Toxicol 32:233–243
Shuang L, Xu F, Jing F, Shu L (2015) Protective roles of selenium on nitric oxide and the gene expression of inflammatory cytokines induced by cadmium in chicken splenic lymphocytes. Biol Trace Elem Res 168:252–260
Sun X et al (2017) Inhibition of bone formation in rats by aluminum exposure via Wnt/β-catenin pathway. Chemosphere 176:1–7
Walton JR (2012) Aluminum disruption of calcium homeostasis and signal transduction resembles change that occurs in aging and Alzheimer’s disease. J Alzheimers Dis (JAD) 29:255–273
Wang H, Li S, Teng X (2015) The antagonistic effect of selenium on lead-induced inflammatory factors and heat shock proteins mRNA expression in chicken livers. Biol Trace Elem Res 171:437–444
Wang Y et al (2016) Significant beneficial association of high dietary selenium intake with reduced body fat in the CODING Study. Nutrients 8:24
Xiang LR et al (2017) The supranutritional selenium status alters blood glucose and pancreatic redox homeostasis via a modulated selenotranscriptome in chickens (Gallus gallus). RSC Adv 7:24438–24445
Zhang C et al (2017) Selenium triggers Nrf2-mediated protection against cadmium-induced chicken hepatocyte autophagy and apoptosis. Toxicol In Vitro 44:349
Zhao X, Yao H, Fan R, Zhang Z, Xu S (2014) Selenium deficiency influences nitric oxide and selenoproteins in pancreas of chickens. Biol Trace Elem Res 161:341–349
Zheng C et al (2016) Aluminum chloride induces neuroinflammation, loss of neuronal dendritic spine and cognition impairment in developing rat. Chemosphere 151:289–295
Zhu X, Jiang M, Song E, Jiang X, Yang S (2015) Selenium deficiency sensitizes the skin for UVB-induced oxidative damage and inflammation which involved the activation of p38 MAPK signaling. Food Chem Toxicol 75:139–145
Acknowledgements
This work was supported by Foshan University research star-up project (099/gg07037) and Key Laboratory of preventive veterinary medicine in Guangdong Provincial Department of Education (2014KTSPT037).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Rights and permissions
About this article

Cite this article
Cao, C., Li, X., Qin, L. et al. High Selenium Yeast mitigates aluminum-induced cerebral inflammation by increasing oxidative stress and blocking NO production. Biometals 31, 835–843 (2018). https://doi.org/10.1007/s10534-018-0128-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-018-0128-0