
Abstract
Manganese (Mn) is an essential element for biological systems, nevertheless occupational exposure to high levels of Mn can lead to neurodegenerative disorders, characterized by serious oxidative and neurotoxic effects with similarities to Parkinson’s disease. The aim of this study was to investigate the potential effects of silymarin (SIL), an antioxidant flavonoid, against manganese chloride induced neurotoxicity both in vivo (cerebral cortex of rats) and in vitro (Neuro2a cells). Twenty-eight male Wistar rats were randomly divided into four groups: the first group (C) received vehicle solution (i.p.) served as controls. The second group (Mn) received orally manganese chloride (20 mg/ml). The third group (Mn + SIL) received both Mn and SIL. The fourth group (SIL) received only SIL (100 mg/kg/day, i.p.). Animals exposed to Manganese chloride showed a significant increase in TBARS, NO, AOPP and PCO levels in cerebral cortex. These changes were accompanied by a decrease of enzymatic (SOD, CAT, GPx) and non-enzymatic (GSH, NpSH, Vit C) antioxidants. Co-administration of silymarin to Mn-treated rats significantly improved antioxidant enzyme activities and attenuated oxidative damages observed in brain tissue. The potential effect of SIL to prevent Mn induced neurotoxicity was also reflected by the microscopic study, indicative of its neuroprotective effects. We concluded that silymarin possesses neuroprotective potential, thus validating its use in alleviating manganese-induced neurodegenerative effects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mn is an essential trace element required for normal growth, development, cellular homeostasis and many ubiquitous enzymatic reactions involved in the neurotransmitter synthesis and metabolism (Aschner and Aschner 2005; Pine et al. 2005; Golub et al. 2005; Dobson et al. 2004). Although small amounts of Mn are a nutritional necessity for normal brain functioning, it has been considered as an environmental neurotoxic at high doses (HaMai and Bondy 2004; Mergler et al. 1999; Pamphlett et al. 2001). This metal is widely used in pesticide formulations (Belpoggi et al. 2002), in water purification as bactericide and fungicide agents and as an antiknock agent in gasoline (Zayed et al. 1999). According to previous findings, excess exposure to high levels of Mn in occupational or environmental settings and therapeutic or disease conditions (Krieger et al. 1995; Spahr et al. 1996; Layrargues et al. 1998) leads to excessive Mn accumulation in the nervous system (Erikson et al. 2007). Thus induces a decrease in dopamine (DA) levels and cell death, syndrome commonly referred to manganism (Barbeau 1984; Sloot et al. 1994).
The cellular and molecular mechanisms of Mn neurotoxicity are not well understood. Generally, Mn is alleged to exert cellular toxicity via a number of mechanisms, including a direct or an indirect formation of reactive oxygen species (ROS) (Ali et al. 1995; Brouillet et al. 1993; Milatovic et al. 2007), oxidation of biological molecules (Archibald and Tyree 1987), and the disruption of Ca2+ and iron homeostasis (Gavin et al. 1990; Kwik-Uribe et al. 2000; Zheng and Zhao 2001). An imbalance between ROS generation and the antioxidant defense mechanisms with subsequent oxidative stress (Betteridge 2000) can initiate apoptosis and/or necrosis in several tissues (Orrenius et al. 2007). Oxidative stress is in fact one of the putative mechanisms by which Mn induces neuronal damages (Dukhande et al. 2006). Efforts have been made to minimize the severity of manganese toxicity via its enhanced sequestration and elimination using different agents. Considering the relationship between manganese exposure and oxidative stress, it is reasonable that administration of some antioxidant and natural biomolecules should be an important therapeutic approach in manganese intoxication. To our knowledge, the effect of silymarin against Mn-toxicity constitutes nowadays the first study.
Silymarin (SIL), is one of the most frequently studied bioflavonoid in the class of flavonols. It is a polyphenolic flavonoid antioxidant isolated from the fruits and seeds of milk thistle Silybum marianum (L.) containing a mixture of several flavonolignans such as silibinin, isosilibinin, silidianin and silichristin (Valenzuela and Garrido 1994). Silymarin has been used clinically in alcoholic disease (Saller et al. 2001). It is also used against various hepatotoxicants including carbon tetrachloride, concanavalin A (Schümann et al. 2003) and aflatoxin B1 (Rastogi et al. 2000). Various studies also indicate that silymarin exhibits cancer preventive effects (Zi et al. 1998; Bhatia et al. 1999). Moreover, silymarin possess number of additional biological effects, such as an antioxidative activity (Valenzuela et al. 1986), anti-inflammatory effects (Manna et al. 1999) and an inhibitory action on tumor necrosis factor-α (TNFα) expression (Zi et al. 1997). Although, a number of compounds, such as trolox and N-acetylcysteine have been used to prevent manganese-induced toxicity in vitro and in vivo (Marreilha dos Santos et al. 2008; Hazell et al. 2006), no studies yet were interested to evaluate the protective effects of silymarin antioxidant flavonoid against Mn-induced neurotoxicity and brain damages. Thus, the aim of this study was to determine first whether changes in the activities of antioxidant enzymes occurred in the cerebral cortex of rats exposed to manganese and second to determine if such changes were associated with oxidative stress by means of lipid peroxidation, protein oxidation and peroxide hydrogen analysis. Furthermore, we have also evaluated whether co-administration of silymarin could modulate these parameters, in vivo and in vitro, and thereby rendered protection to the brain during manganese exposure.
Materials and methods
Chemicals and reagents
Silymarin and all other chemicals, required for all biochemical assays, were obtained from Sigma Chemicals Co. (St. Louis, France).
Animals
Twenty-eight male rats of Wistar strain weighing 190 ± 23 g were obtained from Central Pharmacy (SIPHAT, Tunisia). They were fed pellet diet, purchased from the Industrial Society of rodents diet (SICO, Sfax, Tunisia). Diet composition was detailed by Fetoui et al. (2006, 2007). All animal procedures were conducted in strict conformation with the local Institute Ethical Committee Guidelines for the Care and Use of laboratory animals of our Institution.
Experimental design
After 1 week of acclimatization in a room with controlled temperature (22 ± 3°C) and lighting (12-h light/dark cycle), rats were randomly divided into four groups of seven animals each: The first group of rats served as the control, received ad libitum distilled water and 0.5 ml vehicle solution of silymarin given daily by intraperitoneal (i.p.) injection. The second group (Mn) received, through drinking water, a solution of manganese chloride (20 mg/ml corresponding to 100 mM of Mn2+ (Calabresi et al. 2001; Morello et al. 2007). Animals in the third group (Mn + SIL) were given a single i.p. injection of silymarin (100 mg/kg bw per day), 24 h after the administration of manganese chloride solution. The dose of silymarin used in our study and others (Mansour et al. 2006) gave good protection against the toxicity. The fourth group (SIL) was given daily a single dose of silymarin (100 mg/kg bw). At the end of the experimental period (30 days), the animals in different groups were sacrificed by cervical decapitation to avoid stress conditions. The brain tissue was immediately removed and dissected over ice-cold glass slides and cerebral cortex region was collected and homogenized (10%, w/v) in appropriate phosphate buffer saline (0.1 M, pH 7.4) and centrifuged at 10,000×g for 15 min at 4°C. The resulting supernatants were used for immediate lipid peroxidation and protein oxidation assays. Homogenate aliquots were stored at −80°C for further biochemical assays.
Cell culture and peroxide hydrogen production
Cell culture
Neuro-2a mouse neuroblastoma cell line, obtained from the ATCC (CCL-131), was routinely grown in 75 cm2 flasks (Nunc, Denmark) and maintained in minimum essential medium (MEM) (Invitrogen, Glasgow, UK) supplemented with 10% foetal bovine serum (FBS, Hyclone, Logan, UT), 100 units/ml penicillin and 100 μg/ml streptomycin. Cultures were maintained in a humidified atmosphere with 5% CO2 at 37°C. Cell dissociation was achieved with 0.05% trypsin-0.02% EDTA. Briefly, cells were seeded on 24-well culture plates in medium at an approximate density of 105 cells/cm2 and, after 24 h stabilization, neuronal cells were co-cultured with medium containing various concentrations of MnCl2 (200 and 800 μM) and silymarin (10, 50 and 100 μM) for 24 h. The concentration of MnCl2 was selected based on previously reported cytotoxic levels in cultured cells (dos Santos et al. 2010). For stock solution, MnCl2 was dissolved in MilliQ Plus sterilized water at the concentration of 800 mM and silymarin was dissolved in dimethylsulfoxide (DMSO) to obtain a 100 mM. The experimental concentrations were freshly prepared in the basal medium with a final DMSO concentration of 0.1%.
Measurement of H2O2
Measurement of H2O2 was carried out by the ferrous ion oxidation xylenol orange (FOX1) method (Ou and Wolff 1996). The FOX1 reagent consisted of 25 mM sulphuric acid, 250 μM ferrous ammonium sulfate, 100 μM xylenol orange and 0.1 M sorbitol. Briefly, 100 μl of culture medium were added to 900 μl of FOX1 reagent vortexed and incubated during 30 min at room temperature. Solutions were then centrifuged at 12000×g for 10 min, the amount of H2O2 in the supernatants was determined using spectrophotometer at 560 nm.
Biochemical assays
Enzymatic antioxidants
Catalase (CAT) activity was assayed by the decomposition of hydrogen peroxide according to the method of Aebi (1984). A decrease in absorbance due to H2O2 degradation was monitored at 240 nm for 1 min and the enzyme activity was expressed as μmol H2O2 consumed/min/mg protein.
Total superoxide dismutase activity (SOD) was evaluated by measuring the inhibition of pyrogallol activity as described by Marklund and Marklund (1974). One unit (U) corresponded to the enzyme activity required to inhibit the half of pyrogallol oxidation. SOD activity was expressed as U/mg protein.
Glutathione peroxidase activity (GPx) was measured according to Flohe and Gunzler (1984). The enzyme activity was expressed as nmoles of GSH oxidized/min/mg protein.
Non-enzymatic antioxidants
Acid ascorbic (AA) content was determined spectrophotometrically by dinitrophenyl-hydrazine method described by Jacques-Silva et al. (2001). Briefly, the ascorbic acid in the homogenate was oxidized by Cu2+ to form dihydro-ascorbic acid, which reacts with acidic 4-dinitrophenyl hydrazine to form a red hydrazone. Final color development was achieved with 65% sulfuric acid which was measured at 540 nm. The calibration curve was prepared using ascorbic acid as standard. Results were expressed as μmol/g tissue.
Glutathione (GSH) in tissue was determined by the method of Ellman (1959) modified by Jollow et al. (1974) based on the development of a yellow colour when 5,5-dithiobis-2-nitrobenzoic acid (DTNB) was added to compounds containing sulfhydryl groups. 500 μl of tissue homogenate in phosphate buffer were added to 3 ml of 4% sulfosalicylic acid. The mixture was centrifuged at 1600×g for 15 min. 500 μl of supernatant were taken and added to Ellman’s reagent. The absorbance was measured at 412 nm after 10 min. total GSH content was expressed as μmol/g tissue.
Nitric oxide production was determined based on the Griess reaction (Green et al. 1982). Briefly, 100 μl of deproteinized sample was incubated with 100 μl of the Griess reagent at room temperature for 10 min. Absorbance was measured at 550 nm using a microplate reader. Nitrite concentration was determined from a standard nitrite curve generated using NaNO2. The results were expressed as μM/mg protein.
Lipid peroxidation assay
Lipid peroxidation in the brain tissue was estimated colorimetrically by measuring thiobarbituric acid reactive substances (TBARS) which were expressed in terms of malondialdehyde content according to Draper and Hadley (1990) method. Briefly, 1 ml of cold 5% trichloroacetic acid was added to 500 μl of pre-treated cerebral cortex supernatants and centrifuged at 4000×g for 10 min. 500 μl of the supernatants were transferred to a Pyrex tube and incubated with 1 ml of thiobarbituric acid reagent (TBA, 0.67%) on a boiling water bath for 15 min. The tubes containing the mixture were allowed to cool on running tap water for 5 min. The resulting pink-stained TBARS were determined in a spectrophotometer at 532 nm. The MDA values were calculated using 1,1,3,3-tetraethoxypropane as the standard and expressed as nmoles of MDA/g of tissue.
Protein oxidation assays
Protein carbonyl contents were detected by the reaction with 2,4-dinitrophenylhydrazine (DNPH) method as reported by Evans et al. (1999). Briefly, the DNPH reaction proteins were precipitated with an equal volume of 20% (w/v) trichloroacetic acid and washed three times with 2 ml of an ethanol/ethyl acetate mixture (1:1). Finally, the precipitates were dissolved in 6 M guanidine HCl solution. The absorbance was measured at 370 nm, using the molar extinction coefficient of DNPH, e = 22000 M−1 cm−1 and the results were expressed as nmol of carbonyl/mg protein.
Non-protein thiol (NpSH) levels were determined by the method of Sedlak and Lindsay (1968). A 500 μl aliquot of supernatant was mixed with 10% trichloroacetic acid (V/V). After centrifugation, the protein pellet was discarded and free –SH groups were determined in a clear supernatant. A 100 μl aliquot of supernatant was added to 850 μl of 1 M potassium phosphate buffer (pH 7.4) and 50 μl of DTNB (10 mM). The colorimetric reaction was measured at 412 nm. Reduced glutathione was used as standard. NpSH levels were expressed as μmol NpSH/g tissue.
Advanced protein oxidation products (AOPP) assay was performed by the modification of Witko-Sarsat method (Witko et al. 1992). Briefly, samples were prepared in the following way: 200 μl of the homogenate supernatant fraction was diluted (1:5 v/v) in PBS, 10 μl of 1.16 M potassium iodide were then added to each tube, followed 2 min later by 20 μl acetic acid. AOPP concentrations were expressed as micromoles per liter of chloramine-T equivalents.
Determination of acetylcholinesterase activity
AChE activity was determined after the hydrolysis of acetylthiocholine according to the method of Ellman et al. (1961), modified by Tsakiris et al. (2000). The incubation mixture (1 ml) contained 50 mM Tris–HCl, pH 8, 240 mM sucrose and 120 mM NaCl. The protein concentration of the incubation mixture was 80–100 μg/ml. The reaction was initiated after addition of 0.03 ml of DTNB and 0.05 ml of acetylthiocholine iodide, which was used as substrate. The final concentrations of DTNB and substrate were 0.125 and 0.5 mM, respectively. The increase of absorbance (∆OD) was determined spectrophotometrically at 412 nm.
Quantitative protein determination
Protein concentration in cerebral cortex homogenates were measured by Bradford method (Pierce, BCA Protein Assay Kit, USA) using bovine serum albumin as standard.
Histological studies
Cereberal cortex tissue, extracted from the control and treated rats was fixed in 10% buffered formalin and was processed for paraffin sectioning. Sections of about 5 μm thickness were stained with hematoxylin and eosin to examine under light microscope.
Statistical analysis
The data were analyzed using the statistical package program Stat view 5 Soft Ware for Windows (SAS Institute, Berkeley, CA). Statistical analysis was performed using one-way analysis of variance (ANOVA) followed by Fisher’s protected least significant difference (PLSD) test as a post hoc test for comparison between groups [treated groups (Mn, Mn + SIL, SIL) vs. (controls)] and [Mn + SIL] vs. [(Mn, SIL)]. All values were expressed as mean ± SD. Differences were considered significant if P < 0.05.
Results
Lipid peroxidation product, nitrite formation and protein oxidation on cerebral cortex
The lipid peroxidation product (MDA), nitrite formation (NO), protein carbonyls (PCO) and advanced oxidation protein products (AOPP) in the cerebral cortex are shown in Table 1. A significant increase in MDA (F = 74.91, P < 0.0001), NO (F = 53. 11, P < 0.0001), PCO (F = 10.37, P < 0.006) and AOPP (F = 20.58, P < 0.0001) levels were observed in the cerebral cortex of Mn-treated group compared with the control group. Co-administration of Silymarin at 100 mg/kg significantly decreased (P < 0.01) the level of lipid peroxidation, nitrite formation and protein oxidation products compared with the Mn-treated group. Further, there were no alterations in the lipid peroxidation, nitrite and protein oxidation levels due to silymarin treatment as compared to control group.
Antioxidant enzyme activities in cerebral cortex
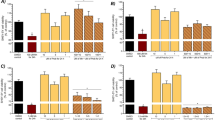
The effects of manganese treatment on the activity of SOD, CAT and GPx in cerebral cortex are shown in Fig. 1. SOD, CAT and GPx activities in cortex were significantly decreased (F = 47.08, P < 0.0001; F = 13.47, P < 0.0001; F = 12.48, P < 0.0002, respectively) upon Mn treatment. Co-treatment with silymarin at 100 mg/kg ameliorated the decreasing activities of these enzymes obtained in the group treated only with Mn.

Antioxidant enzyme activities (CAT, GPx and SOD) in cerebral cortex of controls (C) and rats treated with Manganese chloride (Mn), silymarin (SIL) or their combination (Mn + SIL). a Catalase (CAT). b Superoxide dismutase (SOD). c Glutathione peroxidase (GPx). Values are expressed as means ± SD of six rats in each group. Mn; Mn + SIL and SIL groups vs. control group: * P < 0.05; ** P < 0.01; *** P < 0.001. Mn + SIL group vs. Mn group: ### P < 0.001, ## P < 0.01
Non-enzymatic antioxidant levels in cerebral cortex
Table 2 reported the changes of some non-enzymatic antioxidant parameters in the cortex.
Mn treatment led to a significant decrease in GSH (F = 32.04, P < 0.0001), ascorbic acid (F = 8.82, P < 0.001), Non-protein thiol (F = 18.95, P < 0.0001) levels in the cerebral cortex compared with control group. These effects were significantly mitigated by 100 mg/kg silymarin co-treatment compared with Mn-treated group. These biochemical variables did not differ noticeably between control and silymarin treated groups.
Acetylcholine esterase (AchE) activity
Figure 2 demonstrated the activity of AChE in the cortex of control and treated groups.

Acetylcholinesterase (AChE) activity in cerebral cortex of controls (C) and rats treated with Manganese chloride (Mn), silymarin (SIL) or their combination (Mn + SIL). Values are expressed as means ± SD of six rats in each group. Mn; Mn + SIL and SIL groups vs. control group: * P < 0.05, ** P < 0.01, *** P < 0.001. Mn + SIL group vs. Mn group: # P < 0.05, ## P < 0.01, ### P < 0.001
Acetylcholine esterase activity in the cerebral cortex was significantly inhibited (F = 18.68, P < 0.0001) in Mn-treated group compared to control group. While it reached control values by co-administration of silymarin.
Hydrogen peroxide production in vitro
Figure 3 showed the effects of MnCl2 and silymarin on H2O2 production in Neuro2a Cells. To check if the combination of MnCl2 and silymarin had any benefits, cells were treated with MnCl2 (200 and 800 μM) and varying doses of silymarin (10, 50 and 100 μM) for 24 h. The levels of H2O2 generated in medium of cells were significantly (P < 0.05) increased by 159 and 241% compared to controls after exposure to MnCl2 (200 and 800 μM) respectively, and were significantly (P < 0.05) deceased by (27%, 41% and 20%) cells co-culture with silymarin (10, 50 and 100 μM) and MnCl2 at dose (200 μM) and by (40%, 49% and 25%) at dose (800 μM) compared with MnCl2 alone (200 and 800 μM) respectively.

Effect of SIL on H2O2 production in the medium culture of Neuro2a cells exposed to MnCl2. The cells were exposed to MnCl2 or co-exposed to MnCl2 (200 and 800 μM) and SIL (10, 50 and 100 μM) for 24 h. Data represent the mean ± SD from four independent experiments. * P < 0.05 vs. control, and # P < 0.05 vs. MnCl2 exposed cells
Histological examination of cerebral cortex
Figure 4 illustrates the histopathological assessments of cortex brain tissue in experimental rats. Histopathological examination of the cerebral cortex tissue revealed that manganese treatment caused abnormal cellular arrangement with few pyknotic nucleus, vacuolated spaces and haemorrhage. However, co-administration of silymarin at 100 mg/kg bw prevented these changes and maintained normal architecture with less number of pyknotic nuclei and showed almost normal architecture similar to that of the untreated control. There were no histological alterations in the cerebral cortex of positive controls treated with silymarin alone when compared to negative controls.

Photographs showing histopathological changes in cerebral cortex in different groups, Control group(C), Mn-treated group (Mn), silymarin treated group (SIL), manganese chloride + silymarin treated group (Mn + SIL) (hematoxylin and eosin staining, 400×). ( ) Haemorrhage; (
) Haemorrhage; ( ) vacuolated cytoplasm; (
) vacuolated cytoplasm; ( ) pyknotic nuclei (PN)
) pyknotic nuclei (PN)
Discussion
Human exposure to Mn is of growing concern given its ubiquitous nature and prevalence both in the environment and occupational settings. A recent study suggests that high levels of Mn in drinking water (>300 mg/l) are associated with reduced intellectual function (Wasserman et al. 2006) and induced neurological disorders similar to Parkinson diseases (Aschner 1997; Lander et al. 1999). Recent studies suggest that oxidative stress may play a key role in manganese-induced neurotoxicity (Aschner 1997; Galvani et al. 1995). Therefore the brain is very susceptible to oxidative stress due to its high oxygen consumption, its high iron and lipid contents, especially polyunsaturated fatty acids (PUFA), and the low activity of antioxidant defenses, a fact that makes this tissue more vulnerable to increased levels of oxygen reactive species (Halliwell and Gutteridge 2007). In fact, oxidative stress has been implicated in the pathophysiology of common neurodegenerative disorders, such as Parkinson’s disease, Alzheimer’s disease, as well as in epileptic seizures and demyelination (Bogdanov et al. 2001; Behl and Moosmann 2002; Berg and Youdim 2006).
In the present study, exposure rats to manganese through drinking water resulted in a significant increase in lipid peroxidation, nitrite formation and protein oxidation as indicated by the significant increase in MDA content, NO2 − levels, protein carbonyls and AOPP levels suggesting that Mn activated the formation of free radicals in brain tissue. Our results corroborated with previous findings which demonstrated that Mn exposure stimulated the generation of reactive oxygen species (ROS) (HaMai et al. 2001; Gunter et al. 2006) and enhanced quinones and oxidative species (Migheli et al. 1999; Shen and Dryhurst 1998). Moreover, Milatovic et al. (2009) demonstrated recently that Mn increased F2-isoprostanes formation (lipid peroxidation products) and activated the depletion of ATP in neuronal culture. Although the specific molecular targets that by which Mn-induced oxidative stress are not known, it has been reported that Mn could interact directly with low molecular thiols oxidizing them to disulfides. In fact, reduced cysteinyl residue from proteins could also react with the manganese, which might cause the loss of enzyme catalytic activities (Prabhakaran et al. 2009). The decrease of glutathione, non protein thiols and ascorbic acid contents in cerebral cortex of rats observed in our study supports these findings. Glial cells are also known to protect neurons against oxidative stress and cell death by releasing GSH extracellularly and keeping it in the reduced form (Sagara et al. 1993; Stone et al. 1999). Considering that GSH is the major naturally occurring non-enzymatic antioxidant in the brain (Lissi et al. 1995; Halliwell and Gutteridge 1999; Evelson et al. 2001) this may be related to our results showing that the manganese causes a depletion of sulfhydryl groups, reducing the non-enzymatic antioxidant defenses in cerebral cortex of rats. It is therefore presumed that GSH levels were reduced intracellularly due either to the excess of free radical formation, including NO or to its derivative peroxynitrite forming nitrosoglutathione or by regenerating the nitrosyl groups in order to limit NO deleterious effects (Stamler and Toone 2002; Rodríguez-Martín et al. 2002).
Another explanation of Mn toxicity can also be related to the capacity of this metal to bind transferrin (iron binding protein in blood) and thus affects the binding of Fe to proteins. So the concentration of free intracellular Fe increases and facilitates the Fenton reaction leading to the peroxidation of membrane lipids. Co-administration of silymarin at dose of 100 mg/kg bw reduces significantly lipid peroxidation, nitrite formation and protein oxidation in brain tissue of animals exposed to manganese. We suggest that silymarin scavenges free radical generation by Mn. These effects may reflect the ability of silymarin (i) to enhance the scavenging and inactivation of H2O2 and hydroxyl radicals; (ii) to chelate with redox metals including Fe2+ which catalyzes the formation of free radicals via the Fenton reactions; (iii) and to achieve lipid peroxidation by induction of enzymatic and non-enzymatic antioxidants, such as GSH, SOD and CAT (Miquel et al. 2002). Accordingly, the protection afforded by silymarin against Mn-induced ROS (e.g. H2O2) generation is likely attributable to its antioxidant effects. To confirm this hypothesis, we have also investigated in vitro the neuroprotective properties of silymarin on N2a cells exposed to MnCl2. In this context, cloned neuroblastoma cell lines, including mouse neuroblastoma cell lines (e.g. Neuro2a and N1E-115) and human neuroblastoma cell lines (e.g. SH-SY5 and SK-N-AS) were the established in vitro models that have widely used to investigate the neurotoxicity of xenobiotics. In the present study, exposure of N2a cells line to Mn Cl2 (200 and 800 μM) increased significantly H2O2 production in extracellular medium indicating the role of ROS generation as primary mechanism for Mn-induced toxicity. The ability of SIL to exert great effect on Mn-induced cellular injury is consistent with its increased potency in reducing ROS (e.g. H2O2) generation. Our results are consistent with those of Fu et al. (2009) who have found that silymarin, flavonolignans isolated from S. marianum, has an antioxidative activity and free radical scavenging properties in vitro, which can scavenge various oxidizing radicals such as OH•, NO2 •, O2 •, RNS•. Furthermore, Nencini et al. (2007) demonstrate the efficacity of silymarin in restoring GSH content in brain against acetaminophen-induced neuronal damages. Under physiological conditions, SOD is an important intracellular antioxidant which catalyses the conversion of the superoxide anion radical to molecular oxygen and hydrogen peroxide H2O2 and thus protects against superoxide-induced damage (Hunt et al. 1990). The present study demonstrates that Mn modifies the activity of the antioxidant enzymes by reducing SOD, CAT and GPx. It should be emphasized that these enzymes represent the first barrier against reactive species and are essential to cell survival (Remacle et al. 1992; Matés et al. 1999; Halliwell 2001). The failure in the antioxidant system corroborated with previous studies which demonstrated that superoxide anion, produced in the mitochondrial transport chain, may catalyze the oxidation of Mn2+ to Mn3+. Thus it led to increase oxidant capacity of this metal (Gunter et al. 2006). It is well known that SOD and CAT own sequential functions in ROS removing, by O •2 dismutation, followed by H2O2 conversion to H2O and O2, respectively. The decrease in the activity of SOD can be attributed to the enhanced superoxide production during manganese metabolism. The superoxide radical also inhibits the activity of catalase (Gupta et al. 2005). Silymarin significantly prevents the alterations in the activities of SOD, CAT and GPx in cerebral cortex probably likely attributable to its antioxidant effects.
Acetylcholine, a neurotransmitter associated with learning and memory, is degraded by the enzyme acetylcholinesterase, which achieves its physiological action. In addition to their role in cholinergic transmission, cholinesterases may also play a role during morphogenesis and neurodegenerative diseases (Reyes et al. 1997; Layer et al. 1987). In the present study, the exposure of rats to Mn significantly decreases AchE activity in the cerebral cortex suggesting the ability of Mn2+ to interfere with the calcium action as a regulator of cell function leading to the inhibition of AchE in cholinergic systems. Co-administration of silymarin, a natural antioxidant, to Mn-treated rats improves AchE activity. It seems that the increase of free radicals, observed in the present experiment, may inhibit the acetyl cholinesterase activity. Our results corroborate with previous findings (Tsakiris et al. 2000) which demonstrated that AChE activity in rat brain was inhibited by free radicals. Furthermore, Histopathological examination of the cerebral cortex tissue reveals that manganese treatment causes abnormal cellular arrangement with few pyknotic nuclei, vacuolated spaces and haemorrhage. However, co-treatment with silymarin prevents these changes and also maintains normal architecture with less number of pyknotic nuclei.
In conclusion, the findings of the present study suggest that manganese administration induces the oxidative damage in the cerebral cortex objectified by an increase of lipid peroxidation, protein oxidation, nitrite formation and depletion of enzymatic and non-enzymatic antioxidant. There is a highly reduced capacity to scavenge free radicals produced in the cerebral cortex in response to Mn-neurotoxicity. Silymarin co-administration leads to a significant attenuation in all these parameters. Acting as an antioxidant, silymarin alleviates the oxidative damage in the cerebral cortex. However further investigations are needed to elucidate the precise mechanism of silymarin protection against Mn-neurotoxicity.
References
Aebi H (1984) Catalase in vitro. Methods Enzymol 105:121–126
Ali SF, Duhart HM, Newport GD, Lipe GW, Slikker W Jr (1995) Manganese-induced reactive oxygen species: comparison between Mn2+ and Mn3+. Neurodegeneration 4:329–334
Archibald FS, Tyree C (1987) Manganese poisoning and the attack of trivalent manganese upon catecholamines. Arch Biochem Biophys 256:638–650
Aschner M (1997) Manganese neurotoxicity and oxidative damage. In: Conner JR (ed) Metals and oxidative damage in neurological disorders. Plenum, New York, pp 77–93
Aschner JL, Aschner M (2005) Nutritional aspects of manganese homeostasis. Mol Aspects Med 26(4–5):353–362
Barbeau A (1984) Manganese and extrapyramidal disorders (a critical review and tribute to Dr. George C. Cotzias). Neurotoxicology 5(1):13–35
Behl C, Moosmann B (2002) Oxidative nerve cell death in Alzheimer’s disease and stroke: antioxidants as neuroprotective compounds. Biol Chem 383:521–536
Belpoggi F, Soffritti M, Guarino M, Lambertini L, Cevolani D, Maltoni C (2002) Results of long-term experimental studies on the carcinogenicity of ethylene-bis-dithiocarbamate (Mancozeb) in rats. Ann NY Acad Sci 982:123–136
Berg D, Youdim MB (2006) Role of iron in neurodegenerative disorders. Top Magn Reson Imaging 17:5–17
Betteridge DJ (2000) What is oxidative stress? Metabolism 49:3–8
Bhatia N, Zhao J, Wolf DM, Agarwal R (1999) Inhibition of human carcinoma cell growth and DNA synthesis by silibinin, an active constituent of milk thistle: comparison with silymarin. Cancer Lett 147(1/2):77–84
Bogdanov MB, Andreassen OA, Dedeoglu A, Ferrante RJ, Beal MF (2001) Increased oxidative damage to DNA in a transgenic mouse of Huntington’s disease. J Neurochem 79:1246–1249
Brouillet EP, Shinobu L, Mc Garvey U, Hochberg F, Beal MF (1993) Manganese injection into the rat striatum produces excitotoxic lesions by impairing energy metabolism. Exp Neurol 120:89–94
Calabresi P, Ammassari-Teule M, Gubellini P, Sancesario G, Morello M, Centonze D, Marfia GA, Saulle E, Passino E, Picconi B, Bernardi G (2001) A synaptic mechanism underlying the behavioural abnormalities induced by manganese intoxication. Neurobiol Dis 8:419–432
Dobson AW, Erikson KM, Aschner M (2004) Manganese neurotoxicity. Ann NY Acad Sci 1012:115128
Dos Santos AP, Milatovic D, Au C, Yin Z, Batoreu MC, Aschner M (2010) Rat brain endothelial cells are a target of manganese toxicity. Brain Res 1326:152–161
Draper HH, Hadley M (1990) Malondialdehyde determination as index of lipid peroxidation. Methods Ezymol 86:421–431
Dukhande VV, Malthankar-Phatak GH, Hugus JJ, Daniels CK, Lai JC (2006) Manganese-induced neurotoxicity is differentially enhanced by glutathione depletion in astrocytoma and neuroblastoma cells. Neurochem Res 31:1349–1357
Ellman GL (1959) Tissue sulfhydryl groups. Arch Biochem Biophys 82:70–77
Ellman GE, Courtney KD, Andersen JV, Featherstone RM (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7:88–95
Erikson KM, Dorman DC, Lash LH, Aschner M (2007) Manganese inhalation by rhesus monkeys is associated with brain regional changes in biomarkers of neurotoxicity. Toxicol Sci 97(2):459–466
Evans P, Lyras L, Halliwell B (1999) Measurement of protein carbonyls in human brain tissue. Methods Enzymol 300:145–156
Evelson P, Travacio M, Repetto M, Escobar J, Llesuy S, Lissi EA (2001) Evaluation of total reactive antioxidant potential (TRAP) of tissue homogenates and their cytosols. Arch Biochem Biophys 388:261–266
Fetoui H, Mahjoubi-Samet A, Jammousi K, Ellouze F, Guermazi F, Zeghal N (2006) Energy restriction in pregnant and lactating rats lowers bone mass of their progeny. Nutr Res 26:421–426
Fetoui H, Mahjoubi-Samet A, Jamoussi K, Ayadi F, Ellouze F, Zeghal N (2007) Food restriction in pregnant and lactating rats induces anemia and increases plasma lipid peroxidation in their progeny. Nutr Res 27:788–793
Flohe L, Gunzler WA (1984) Assays of glutathione peroxidase. Method Enzymol 105:114–121
Fu H, Lin M, Muroya Y, Hata K, Katsumura Y, Yokoya A, Shikazono N, Hatano Y (2009) Free radical scavenging reactions and antioxidant activities of silybin: mechanistic aspects and pulse radiolytic studies. Free Radic Res 43(9):887–897
Galvani P, Fumagalli P, Santagostino A (1995) Vulnerability of mitochondrial complex I in PC12 cells exposed to manganese. Eur J Pharm Sci 293(4):377–383
Gavin CE, Gunter KK, Gunter TE (1990) Manganese and calcium efflux kinetics in brain mitochondria. Relevance to manganese toxicity. Biochem J 266(2):329–334
Golub MS, Hogrefe CE, Germann SL, Tran TT, Beard JL, Crinella FM, Lonnerdal B (2005) Neurobehavioral evaluation of rhesus monkey infants fed cow’s milk formula, sow formula or soy formula with added manganese. Neurotoxicol Teratol 27:615–627
Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR (1982) Analysis of nitrate, nitrite, and [15N] nitrate in biological fluids. Anal Biochem 126:131–138
Gunter TE, Gavin CE, Aschner M, Gunter KK (2006) Speciation of manganese in cells and mitochondria: a search for the proximal cause of manganese neurotoxicity. Neurotoxicology 27:765–776
Gupta R, Kannan GM, Sharma M, Flora SJS (2005) Therapeutic effects of Moringa oleifera on arsenic-induced toxicity in rats. Environ Toxicol Pharmacol 20:456–464
Halliwell B (2001) Role of free radicals in the neurodegenerative diseases. Therapeutic implications for antioxidant treatment. Drugs Aging 18(9):685–716
Halliwell B, Gutteridge JMC (1999) Antioxidant defense enzymes. In: Halliwell B, Gutteridge JMC (eds) Free radicals in biology and medicine, 3rd edn. Oxford University Press, Oxford, pp 107–146
Halliwell B, Gutteridge JMC (2007) Measurement of reactive species. In: Halliwell B, Gutteridge JMC (eds) Free Radicals in Biology and Medicine, 4th edn. Oxford University Press, Oxford, pp 268–340
HaMai D, Bondy SC (2004) Oxidative basis of manganese neurotoxicity. Ann NY Acad Sci 1012:129–141
HaMai D, Campbell A, Bondy SC (2001) Modulation of oxidative events by multivalent manganese complexes in brain tissue. Free Radic Biol Med 31:763–768
Hazell AS, Normandin L, Norenberg MD, Kennedy G, Yi JH (2006) Alzheimer type II astrocytic changes following sub-acute exposure to manganese and its prevention by antioxidant treatment. Neurosci Lett 396(3):167–171
Hunt JV, Smith CC, Wolff SP (1990) Autooxidative glycosylation and possible involvement of peroxides and free radicals in LDL modification by glucose. Diabetes 39(11):1420–1424
Jacques-Silva MC, Nogueira CW, Broch LC, Flores EM, Rocha JBT (2001) Diphenyl diselenide and ascorbic changes deposition of selenium and ascorbic in liver and brain of mice. Pharmacol Toxicol 88:119–125
Jollow DJ, Mitchell JR, Zampaglione N, Gillette JR (1974) Bromobenzene-induced liver necrosis. Protective role of glutathione and evidence for 3,4-bromobenzene oxide as the hepatotoxic metabolite. Pharmacology 11:151–169
Krieger D, Krieger S, Jansen O, Gass P, Theilmann L, Lichtnecker H (1995) Manganese and chronic hepatic encephalopathy. Lancet 346:270–274
Kwik-Uribe CL, Golub MS, Keen CL (2000) Chronic marginal iron intakes during early development in mice alter brain iron concentrations and behavior despite postnatal iron supplementation. J Nutr 130:2040–2048
Lander F, Kristiansen J, Lauritsen JM (1999) Manganese exposure in foundry furnacemen and scrap recycling workers. Int Arch Occup Environ Health 72:546–550
Layer PG, Alber R, Sporns O (1987) Quantitative development and molecular forms of acetylcholinesterase and butyrylcholinesterase during morphogenesis and synaptogenesis of chick brain and retina. J Neurochem 49(1):175–182
Layrargues GP, Rose C, Spahr L, Zayed J, Normandin L, Butterworth RF (1998) Role of manganese in the pathogenesis of portal-systemic encephalopathy. Metab Brain Dis 13:311–317
Lissi E, Salim-Hanna M, Pascual C, del Castillo MD (1995) Evaluation of total antioxidant potential (TRAP) and total antioxidant reactivity from luminal enhanced chemiluminescence measurements. Free Radical Biol Med 18:153–158
Manna SK, Mukhopadhyay A, Van NT, Aggarwal BB (1999) Silymarin suppresses TNF-induced activation of NF-kappa B, c-Jun N-terminal kinase, and apoptosis. J Immunol 163(12):6800–6809
Mansour HH, Hafez HF, Fahmy NM (2006) Silymarin modulates Cisplatin-induced oxidative stress and hepatotoxicity in rats. J Biochem Mol Biol 39:656–661
Marklund S, Marklund G (1974) Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem 47:469–474
Marreilha dos Santos AP, Santos D, Au C, Milatovic D, Aschner M, Batoréu MC (2008) Antioxidants prevent the cytotoxicity of manganese in RBE4 cells. Brain Res 1236:200–205
Matés JM, Pérez-Gómez C, Núñez de Castro I (1999) Antioxidant enzymes and human diseases. Clin Biochem 32:595–603
Mergler D, Baldwin M, Bélanger S, Larribe F, Beuter A, Bowler R, Panisset M, Edwards R, de Geoffroy A, Sassine MP, Hudnell K (1999) Manganese neurotoxicity, a continuum of dysfunction: results from a community based study. Neurotoxicology 20:327–342
Migheli A, Piva R, Casolino S, Atzori C, Dlouhy SR, Ghetti B (1999) A cell cycle alteration precedes apoptosis of granule cell precursors in the weaver mouse cerebellum. Am J Pathol 155:365–373
Milatovic D, Yin Z, Gupta RC, Sidoryk M, Albrecht J, Aschner JL, Aschner M (2007) Manganese induces oxidative impairment in cultured rat astrocytes. Toxicol Sci 98(1):198–205
Milatovic D, Zaja-Milatovic S, Gupta RC, Yu Y, Aschner M (2009) Oxidative damage and neurodegeneration in manganese-induced neurotoxicity. Toxicol Appl Pharmacol 240(2):219–225
Miquel J, Bernd A, Sempere JM, Díaz-Alperi J, Ramírez A (2002) The curcuma antioxidants: pharmacological effects and prospects for future clinical use. A review. Arch Gerontol Geriatr 34:37–46
Morello M, Zatta P, Zambenedetti P, Martorana A, D’Angelo V, Melchiorri G, Bernardi G, Sancesario G (2007) Manganese intoxication decreases the expression of manganoproteins in the rat basal ganglia: an immunohistochemical study. Brain Res Bull 74:406–415
Nencini C, Giorgi G, Micheli L (2007) Protective effect of silymarin on oxidative stress in rat brain. Phytomedicine 14:129–135
Orrenius S, Gogvadze V, Zhivotovsky B (2007) Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol 47:143–183
Ou P, Wolff SP (1996) A discontinuous method for catalase determination at near physiological concentrations of H2O2 and its application to the study of H2O2 fluxes within cells. J Biochem Biophys Methods 31:59–67
Pamphlett R, McQuilty R, Zarkos K (2001) Blood levels of toxic and essential metals in motor neuron disease. Neurotoxicology 22:401–410
Pine M, Lee B, Dearth R, Hiney JK, Dees WL (2005) Manganese acts centrally to stimulate luteinizing hormone secretion: a potential influence on female pubertal development. Toxicol Sci 85(2):880–885
Prabhakaran K, Chapman GD, Gunasekar PG (2009) BNIP3 up-regulation and mitochondrial dysfunction in manganese-induced neurotoxicity. NeuroToxicology 30(3):414–422
Rastogi R, Srivastava AK, Srivastava M, Rastogi AK (2000) Hepatocurative effect of picroliv and silymarin against aflatoxin B1 induced hepatotoxicity in rats. Planta Med 66:709–713
Remacle J, Michiels C, Raes M (1992) The importance of antioxidant enzymes in cellular aging and degeneration. EXS 62:99–108
Reyes AE, Perez DR, Alvarez A, Garrido J, Gentry MK, Doctor BP, Inestrosa NC (1997) A monoclonalantibody against acetylcholinesterase inhibits the formation of amyloid Wbrils induced by the enzyme. Biochem Biophys Res Commun 232(3):652–655
Rodríguez-Martín E, Casarejos MJ, Canals S, de Bernardo S, Mena MA (2002) Thiolic antioxidants protect from nitric oxide-induced toxicity in fetal midbrain cultures. Neuropharmacology 43:877–888
Sagara JI, Miura K, Bannai S (1993) Maintenance of neuronal glutathione by glial cells. J Neurochem 61:1672–1676
Saller R, Meier R, Brignoli R (2001) The use of silymarin in the treatment of liver diseases. Drugs 61:2035–2063
Schümann J, Prockl J, Kiemer AK, Vollmar AM, Bang R, Tiegs G (2003) Silibinin protects mice from T cell-dependent liver injury. J Hepatol 39:333–340
Sedlak J, Lindsay RH (1968) Estimation of total, protein bound, and non-protein sulfhydryl groups in tissue with Ellman’s reagent. Anal Biochem 25:192–205
Shen XM, Dryhurst G (1998) Iron- and manganese-catalyzed autoxidation of dopamine in the presence of l-cysteine: possible insights into iron- and manganese-mediated dopaminergic neurotoxicity. Chem Res Toxicol 11(7):824–837
Sloot WN, van der Sluijs-Gelling AJ, Gramsbergen JB (1994) Selective lesions by manganese and extensive damage by iron after injection into rat striatum or hippocampus. J Neurochem 62:205–216
Spahr L, Butterworth RF, Fontaine S, Bui L, Therrien G, Milette PC, Lebrun LH, Zayed J, Leblanc A, Pomier-Layrargues G (1996) Increased blood manganese in cirrhotic patients: relationship to pallidal magnetic resonance signal hyperintensity and neurological symptoms. Hepatology 24:1116–1120
Stamler JS, Toone EJ (2002) The decomposition of thionitrites. Curr Opin Chem Biol 6:779–785
Stone R, Stewart VC, Hurst RD, Clark JB, Heales SJ (1999) Astrocyte nitric oxide causes neuronal mitochondrial damage, but antioxidant release limits neuronal cell death. Ann NY Acad Sci 893:400–403
Tsakiris S, Angelogianni P, Schulpis KH, Stavridis JC (2000) Protective effect of l-phenylalanine on rat brain acetylcholine esterase inhibition induced by free radicals. Clin Biochem 33(2):103–106
Valenzuela A, Garrido A (1994) Biochemical bases of the pharmacological action of the flavonoid silymarin and of its structural isomer silibinin. Biol Res 27(2):105–112
Valenzuela A, Guerra R, Videla LA (1986) Antioxidant properties of the flavonoids silybin and (þ)-cyanidanol-3: comparison with butylated hydroxyanisole and butylated hydroxytoluene. Planta Med 52(6):438–440
Wasserman GA, Liu X, Parvez F, Ahsan H, Levy D, Factor-Litvak P et al (2006) Water manganese exposure and children’s intellectual function in Araihazar, Bangladesh. Environ Health Perspect 114:124–129
Witko V, Nguyen AT, Descamps-Latscha B (1992) Microtiter plate assay for phagocyte derived taurine-chloramines. J Clin Lab Anal 6:47–53
Zayed J, Hong B, L’Espérance G (1999) Characterization of manganese- containing particles collected from the exhaust emissions of automobiles running with MMT additive. Environ Sci Technol 33(19):3341–3346
Zheng W, Zhao Q (2001) Iron overload following manganese exposure in cultured neuronal, but not neuroglial cells. Brain Res 897:175–179
Zi X, Mukhtar H, Agarwal R (1997) Novel cancer chemopreventive effects of a flavonoid antioxidant silymarin: inhibition of mRNA expression of an endogenous tumor promoter TNF alpha. Biochem Biophys Res Commun 239(1):334–339
Zi X, Feyes DK, Agarwal R (1998) Anticarcinogenic effect of a flavonoid antioxidant, silymarin, in human breast cancer cells MDA-MB 468: induction of G1 arrest through an increase in Cip1/p21 concomitant with a decrease in kinase activity of cyclindependent kinases and associated cyclins. Clin Cancer Res 4(4):1055–1064
Acknowledgments
The authors are indebted to Miss Dalenda Kchaou for their assistance in histolological techniques. The present work was supported by the grants of DGRST (Appui a la Recherche Universitaire de base, ARUB 99/UR/08-73), Tunisia.
Author information
Authors and Affiliations
Corresponding author
Additional information
H. Fetoui and M. Sefi contributed equally to this work.
Rights and permissions
About this article
Cite this article
Chtourou, Y., Fetoui, H., Sefi, M. et al. Silymarin, a natural antioxidant, protects cerebral cortex against manganese-induced neurotoxicity in adult rats. Biometals 23, 985–996 (2010). https://doi.org/10.1007/s10534-010-9345-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-010-9345-x