
Abstract
The widespread occurrence of cadmium in the environment continues to pose a threat to human health despite attempts at limiting its technological uses. The biologically significant ionic form of cadmium, Cd2+, binds to many bio-molecules and these interactions underlie the toxicity mechanisms of cadmium. Some of the molecules specialized in the handling of alkaline earth (Mg2+, Ca2+) and transition metal ions (e.g. Zn2+, Cu2+/+, Fe3+/2+) should be particularly sensitive to the presence of Cd2+, because they enclose cationic sites to which the toxic metal can bind. The possible molecular targets of this kind for cadmium are considered herein. Whereas in vitro evidence for native cation replacement by Cd2+ in bio-molecules has been largely provided, the demonstration of such occurrences in vivo is scarce, with the notable exception of metallothionein. One reason might be that realistic low-level Cd2+ contaminations involve cellular concentrations far smaller than those of endogenous cations that usually saturate their binding sites. It is very likely that cadmium toxicity is most often mediated by biological systems amplifying the signals triggered by the presence of Cd2+. The interference of Cd2+ with redox sensitive systems acting at the transcriptional and post-transcriptional levels is instrumental in such processes. A better understanding of cadmium toxicity to tackle the environmental challenges lying ahead thus requires properly designed studies implementing biologically relevant cadmium concentrations on different cell types, improved knowledge of the homeostasis of essential metals, and use of these data in a theoretical framework integrating all cellular aspects of cadmium effects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cadmium is a widespread metal contaminating many areas, either naturally or because of industrial use (Lane and Morel 2000; Pan et al. 2010). Although some forms of life adjust to the presence of this metal (Morel 2008), most biological effects of cadmium are deleterious, particularly to mammals and other species located at the top of the evolution tree. The modes of exposure to cadmium and its health effects are considered elsewhere, e.g. (Järup and Åkesson 2009; Johri et al. 2010; Pan et al. 2010; Satarug et al. 2010; Nawrot et al. 2010).
Cadmium toxicity has historically been closely associated with zinc homeostasis and oxidative stress in mammalian cells. The reasons for this association are to be found in the protection often afforded by zinc against the deleterious consequences of cadmium exposure, as most recently restated (Jacquillet et al. 2006; El Heni et al. 2009; Rogalska et al. 2009), and in the chemical similarities between the two metals. Experimental evidence for a shift of the cellular redox balance induced by cadmium abounds (Liu et al. 2008; Cuypers et al. 2010), and zinc has well established antioxidant properties (Maret 2008). The cellular redox potential depends on a large number of parameters, but most transition metals bearing unpaired electrons are very efficient catalysts to convert relatively inert species into highly oxidizing compounds (see below Cadmium toxicity and pathways modulating the homeostasis of essential metals). Thus, deregulation of the homeostasis of transition metals by cadmium, and cellular cadmium traffic via pathways dedicated to transition metals contribute to the toxicity mechanisms of cadmium.
The aim of the present review is to examine cadmium toxicity interfering with the homeostasis of essential metal cations in animal cells. The field of metal homeostasis has been booming over the last 10–15 years, and many aspects of it are regularly reviewed in details, as are new developments in cadmium toxicity. They will not be reproduced here. It is hoped that drawing the attention of readers to the complex interplay between cellular handling of essential and toxic metal cations will stimulate work aiming at integrating experimental data into thorough descriptions of the cellular behavior under cadmium stress. Also, general concepts and observations will be considered in priority, and the cellular context in which they are relevant will only be indicated when restrictions are important. Last, mainly biochemical and cellular mechanisms of cadmium toxicity will be presented, setting aside other interesting aspects of cadmium toxicity that can be easily found elsewhere, e.g. (Bhattacharyya 2009; Byrne et al. 2009; Edwards and Prozialeck 2009; Järup and Åkesson 2009; Liu et al. 2009; Hartwig 2010; Nawrot et al. 2010; Johri et al. 2010; Satarug et al. 2010).
Replacement of biological metals by cadmium
An important aspect in exploring the mechanisms of cadmium toxicity is to identify the most likely binding sites for the toxic cation. Among them, the active sites of different metalloproteins are prominent candidates as they are designed to accommodate metal cations (Fig. 1). Zinc and cadmium belong to the same column of the periodic table. Both do not change oxidation states (their d electronic layer is full) and they occur as divalent cations in biological environments. A large diversity of bio-molecules can be ligands of these divalent cations, as functional groups with sulfur, nitrogen, or oxygen binding atoms can all contribute to the first coordination sphere of cadmium. For instance cadmium in metallothionein (MT) is exclusively bound to sulfur, but cadmium coordination is mixed in one zinc site of alcohol dehydrogenase (Fig. 1), or several zinc finger proteins. Also, replacement of calcium ions by cadmium defines a mainly oxygenated environment for the cation (Fig. 1).

Structures of cadmium-substituted active sites of zinc and calcium proteins, and other examples of zinc-binding sites in proteins. LADH: liver alcohol dehydrogenase, native (zinc) and cadmium-substituted catalytic site, Protein Databank ID 1HET and 1HEU, respectively. PARV: native (calcium) and cadmium-substituted carp parvalbumin, PDB ID 5CPV and 1CDP, respectively. Lck: SH3 domain of human tyrosine kinase Lck (2IIM). Note that the two residues on top (His70 and Gly58) belong to different polypeptides than the two (Asp79 and His 76) at the bottom. Cu–ZnSOD: Cu-Zn human superoxide dismutase (2VOA). Atoms color code for central metal ions: zinc, gray; cadmium, magenta; copper, orange; calcium, light gray; for ligand atoms: blue, nitrogen; red, oxygen; green, carbon; yellow, sulfur. This Figure was prepared with the PyMOL software (http://www.pymol.org)
Hydrolysis of water coordinated to cadmium is ca. one order of magnitude faster than in the case of zinc or most other ions of transition metals. This rate constant is similar for cadmium and calcium. The ionic radius of Cd2+ is ca. 20% larger than that of Zn2+, but within 5% of that of Ca2+ in similar environments. All these chemical properties contribute to the dynamics of cadmium trafficking by ligand exchange. The diversity of cadmium ligands implies that many cation binding sites can accommodate Cd2+ under favorable conditions as can be achieved in vitro. Although no strict general rule can be drawn from the available data on proteins, cadmium binding to mainly oxygen/nitrogen sites is generally weaker than binding of the natural cation. This is the case for the angiotensin-converting enzyme 1, for instance, with a two orders of magnitude difference between the apparent affinity constants of zinc and cadmium (Carvalho et al. 1995). When the number of sulfur atoms increases in the coordination sphere, so does the association constant. Demonstration of this trend is given by the variants of a zinc finger consensus peptide: the peptide providing a His2Cys2 coordination sphere to zinc is more stable than with any other metal tested, whereas that with a Cys4 site binds cadmium more tightly. The affinities of zinc and cadmium to the HisCys3 site are similar (Berg and Godwin 1997).
Although many Cd2+-bound species can be obtained in vitro in the presence of large metal excess with pure bio-molecules (Fig. 1), demonstration of their occurrence in vivo, in mammalian cells in particular, is not an easy task. To give a single example among many, the inhibition of the transcription factor p53 by cadmium, and other metals, has been evidenced, including in MCF7 human breast carcinoma cells, and cadmium interacts with the protein (Méplan et al. 1999). However, the actual proof of zinc replacement by cadmium in the protein was not provided, and means to probe this aspect were not implemented by the authors (Méplan et al. 1999, 2000). The conformational change induced by cadmium (and other inhibitory metals) to give the so-called “mutant” conformation may even be taken as a sign of alternative binding for cadmium, since isostructural replacement of zinc at HisCys3 binding sites should not strongly impact folding (Berg and Godwin 1997). Therefore, cadmium does inhibit p53, including in cells (Méplan et al. 1999, 2000), but this may not occur by the strict replacement of zinc. Of particular interest in this respect is the report indicating that p53 is phosphorylated upon exposing MCF7 cells to cadmium (Matsuoka and Igisu 2001). Considering all cadmium substitutions at the metal sites of metalloproteins performed in vitro and the scarcity of data demonstrating such occurrences in vivo, care should be exercised before interpreting cadmium toxicity data with this simple molecular explanation. Yet, cadmium replacement of other metals in cellular proteins do occur, in MT (Margoshes and Vallee 1957) or carbonic anhydrase (Lane and Morel 2000) for instance.
Exposure to cadmium does perturb the homeostasis of other metals, and, reciprocally, cadmium effects depend on the body status for essential metals such as iron and zinc. This is regularly observed in a variety of conditions, e.g. (Nakazato et al. 2008; Wills et al. 2008; Kippler et al. 2009). A survey of the relevance of metal homeostasis to cadmium toxicity follows.
Cadmium toxicity and zinc homeostasis
Schematic overview of zinc involvement in Biology
Although zinc is classified as a trace element in Biology, the number and the diversity of biological molecules with which it forms complexes are amazing. Rough estimates, mainly based on scans of sequenced genomes for potential zinc binding sites in proteins, have given values of zinc-interacting molecules in the thousands (Maret 2009; Maret and Li 2009). Accordingly, the biological functions in which zinc takes part include gene expression, cellular proliferation and differentiation, growth and development, apoptosis, and the immune response to name but a few (Vallee and Falchuk 1993).
Many of these functions rely on the presence of zinc at the active sites of enzymes (Auld 2001), as partly illustrated in Fig. 1. It is the case of the numerous hydrolases, including metalloproteinases, lyases, and dehydrogenases which leave a ‘free’ (actually water/hydroxide bound) coordination position around the zinc atom for substrate binding and catalysis. Zinc may also occur in combination with other metals at the active sites of some enzymes such as superoxide dismutase (SOD) (Fig. 1). Last, other sites with a full coordination sphere for zinc assist in protein folding and maturation, without any direct role in catalysis.
Besides its involvement at the active site of enzymes, zinc has been more recently associated with an increasing number of regulatory functions. Zinc plays a catalytic, inhibitory, or accessory role in regulatory enzymes such as kinases or phosphatases. It is a critical part of zinc-finger domains that regulate scores of protein–protein or protein–nucleic acids interactions. Furthermore, a single zinc cation can bind to amino acid side chains belonging to different proteins, and thus it can act as a cross-linking agent in the assembly of large protein complexes. This occurs when lymphocyte protein tyrosine kinase (Lck) is recruited to the T cell receptor (TCR) complex upon activation, as recently reviewed (Haase and Rink 2009) and illustrated in Fig. 1. Last, some specialized cells transiently secrete large amounts of zinc from acidic vesicles to communicate with nearby cells. For instance, pancreatic β-cells abundantly accumulate zinc, probably via ZnT-8 (SLC30A8), in insulin granules and release them to adjust blood glucose concentrations (Wijesekara et al. 2009). Genome-wide association studies between the ZnT8 rs13266634 SNP and (mainly type 2) diabetes (Jansen et al. 2009) support the role of zinc in insulin regulation. Also, some glutamatergic neurons release zinc in synapses from vesicles filled via ZnT-3 (SLC30A3) (Frederickson et al. 2005). This release regulates several receptors, such as NMDA (N-methyl-D-aspartate)-, GABA (γ-aminobutyric acid)-, or glycine receptors, and transporters and it explains the neuromodulatory action of zinc (Sensi et al. 2009).
From the above summary, it appears that the biological status of zinc has largely shifted over the last few years from an exclusively catalytic/structural element to a significant signaling atom with the role of second messenger like calcium, in brain function in particular (Frederickson and Bush 2001).
Molecular components involved in zinc traffic
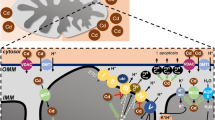
With respect to the wide range of biological functions fulfilled by zinc, progress in the identification of the molecular components responsible for its traffic has been comparatively slow. A schematic view of cellular zinc homeostasis is relatively simple, and most identified participants are likely cadmium targets (Fig. 2).

Schematic representation of zinc homeostasis relevant to cadmium toxicity. Arrows indicate the direction of zinc flow. Targets of cadmium are shown with a Cd symbol within a blue star for those being demonstrated and within a circle for those without in vivo experimental evidence. These associations do not necessarily mean direct interactions between Cd2+ and the indicated target
The initial discovery of the mammalian transporter opposing the zinc-sensitivity of baby hamster kidney cells (Palmiter and Findley 1995) has been followed by the identification of a family of genes encoding ZnT (SLC30). Ten such genes have been found in the human genome and their products, differing by their tissue expression and their sub-cellular localization, exclude zinc from the cytosol (Lichten and Cousins 2009). The mechanism of Zn2+ transport by ZnT remains an open question, but it seems likely that the traffic of the cation through membranes may be coupled with that of other ions such as protons, Ca2+, or Na+ (Sekler et al. 2007; Ohana et al. 2009). No evidence for cadmium transport by any ZnT has been published.
In a mirroring function, another family of membrane proteins is usually associated with zinc import into the cytosol. The human genome contains fourteen genes encoding proteins assigned this function, and many homologous genes are found in other organisms (Eng et al. 1998). These transporters are also involved in the traffic of other metals (Lichten and Cousins 2009). The detailed transport mechanism has not been elucidated for all members, but it does not seem to be coupled to other cations. Rather, anionic (HCO3 −) symport has been proposed for ZIP8 and ZIP14 (He et al. 2009). The role of ZIP transporters in cadmium traffic is considered below under the “Cadmium toxicity and manganese homeostasis” section.
Despite the large number of ZnT and ZIP transporters, different observations indicate that zinc can cross membranes via other proteins, such as voltage- (Gyulkhandanyan et al. 2006) or ligand-gated (Weiss and Sensi 2000) cation channels, among others. Interestingly, the evidenced or suspected transporters for Zn2+ obey different transport mechanisms, whether anti-gradient energy driven, diffusion controlled, or coupled anion- or cation- exchange for instance (Haase and Beyersmann 2002; Sekler et al. 2007). It means that the actual mediators of Zn2+ transfer through membranes most likely vary with the detailed cellular conditions, including the actual Zn2+ concentrations on both sides of the considered membrane, and with the cell type. This variety of potential Zn2+ transporters is another similarity between Zn2+ and Ca2+ homeostasis, and Cd2+ toxicity should be considered for all aspects of Zn2+ and Ca2+ functions.
Intracellular handling of zinc
The large zinc-binding capacity of MT indicates that Zn2+ is extensively chelated in the cytosol (Maret and Krężel 2007). MT were initially assigned a detoxifying role (Hamer 1986), of cadmium in particular, mainly because no data indicating a clear function were rapidly obtained. It now appears that MT are involved in at least two major types of reactions: zinc buffering and zinc exchange between proteins (Maret and Krężel 2007). Furthermore, increasing intracellular zinc concentrations, and the ensuing increase in MT production, seem to be associated with partial translocation of the loaded protein to the nucleus (Chimienti et al. 2001). The function of MT redistribution upon heavy zinc load and in other situations (Beyersmann and Haase 2001) is not elucidated, but it may provide another level of regulation of the intracellular zinc concentration. Considering the tight binding of Cd2+ by MT and the sensitivity of the expression of MT genes to stressful conditions, these proteins may mediate cadmium toxicity in various ways (Sabolić et al. 2010), including by decreasing the zinc buffering ability of cells in different compartments, by changing the dynamics of zinc exchanges, and by decreasing the cellular antioxidant defense.
Although metal regulation is widespread throughout all forms of life, only one transcription factor is believed to be directly (i.e. by direct binding) responsive to a transition metal in mammalian cells. This transcription factor is MTF-1 (metal-responsive-element-binding transcription factor-1), a large multidomain-protein with a series of six zinc-finger modules in the DNA binding domain and another part with transcriptional activation domains (Laity and Andrews 2007). MTF-1 (up)regulates a range of genes, with those encoding MT and ZnT-1 prominent among them (Lichtlen et al. 2001), and it is activated by different stimuli including, but not restricted to, high metal (zinc, cadmium, or copper) concentrations. Zinc (Laity and Andrews 2007) and copper (Chen et al. 2008) ions have been shown to directly bind to MTF-1. Cadmium binding to the zinc finger domains, with zinc displacement, seems possible, although it has not been formally demonstrated in cells. But cadmium activation of MTF-1 may use other mechanisms (Saydam et al. 2002; Zhang et al. 2003), as reviewed in (Martelli et al. 2006). Indeed, if cellular conditions are such that Cd2+ expels Zn2+ from MT or if any of these cations activates transduction pathways regulating MTF-1, activation of the latter transcription factor may appear as a direct consequence of cadmium exposure. The complexity of MTF-1 regulation (Lindert et al. 2009) leaves room for a diversity of mechanisms.
Intracellular zinc stores
None of the known zinc-binding proteins can be considered a storage protein. The 7:1 stoichiometry of Zn:MT is on the upper limit of the specific binding capacity of proteins, and, although many bio-molecules may display non-specific zinc binding sites, no evidence for heavily zinc-decorated proteins has been produced up to now. Hence the large intracellular reservoirs of zinc, that seem mandatory for some functions such as massive excretion from zinc vesicles (see Schematic overview of zinc involvement in Biology), are ill-defined. The sub-cellular zinc distribution spreads over different organelles (Fig. 2) and it is in dynamic equilibrium (Haase and Beyersmann 2002). Few comparative studies between zinc and cadmium distributions are available for mammalian cells. Using fluorescent probes for these metals, both Cd2+ and Zn2+ seem to be distributed similarly. For instance, 5-nitrobenzothiazole coumarin (BTC-5N), a fluorescent chelator of both of these cations, affords similar patterns for each of them with clear perinuclear spots, and an increase of the fluorescent intensity with the amount of added cadmium (Rousselet et al. 2008b). Such experiments indicate that BTC-5N has access to cations located in similar places, but it is not clear if all forms of the metals are visualized with such fluorescent probes. Therefore, easily mobilized Cd2+, i.e. the fraction displaced by the probe, overlaps with easily mobilized Zn2+, but the consequences of accumulated Cd2+ ions on the distribution and dynamics of exchange of the intracellular Zn2+ pools is not known. This open question should be the topic of further studies.
Cadmium toxicity and iron homeostasis
Salient features of iron homeostasis relevant to cadmium toxicity
The knowledge gained about mammalian iron homeostasis has been tremendous over slightly more than a decade. The field is regularly covered by frequent reviews, and only features that can be predicted or have been demonstrated to be relevant for cadmium toxicity are recalled here (Fig. 3).

Schematic representation of iron homeostasis relevant to cadmium toxicity. The symbols and cautious note of the legend of Fig. 2 are also valid for this figure. Dotted arrows point to the proteins, the mRNA of which are regulated by Iron Regulatory Proteins. Abbreviations: FPN, ferroportin; FTH1-FTL, FT subunits; Tf, transferrin; TfR1, transferrin receptor; Hepc, hepcidin
By far, the prominent role of iron in mammalian biology is to convey oxygen to tissues to fuel the cellular machinery. Oxygen is bound to hemoglobin and myoglobin, and erythrocytes are responsible for its proper distribution throughout the body. Anemia is among the various symptoms of cadmium intoxication (Järup 2003; Nordberg 2009), albeit as a relatively late sign after the onset of cadmium exposure (Rousselet and Moulis 2008). Erythropoiesis is a complex and gradual process requiring many different steps and associated molecular mechanisms, from progenitors to mature red blood cells in the bone marrow (Tsiftsoglou et al. 2009). A major erythropoiesis-stimulating agent is erythropoietin, a mainly renal-produced and hypoxia-promoted hormone (Fried 2009). Kidney is a target organ of cadmium toxicity, and erythropoietin synthesis is decreased by accumulation of the metal (Horiguchi et al. 2000). Iron is also found at the active site of many proteins and enzymes involved in functions that are critical for life. Therefore mammals, and most other forms of life, have developed sophisticated means to ensure provision of proper iron concentrations.
Cadmium traffic and iron transport
In contrast to zinc that does not seem to be driven to a specific organelle for biosynthetic and other needs, a majority of cellular iron has to reach mitochondria for heme and iron-sulfur clusters biogenesis (Fig. 3). A breakthrough in iron homeostasis was the discovery of the transporter responsible for iron absorption (Gunshin et al. 1997). This membrane protein (DCT1, DMT1, Nramp2, or SLC11A2) is likely to be the main mediator of iron entry for cells exposed to external sources of iron, such as enterocytes or pneumocytes (Garrick and Garrick 2009). DMT1 is a proton symport which displays optimal transport activity at low pH. DMT1 is not a very selective gate for iron, and absorption of other cations, including cadmium, via this transporter has been put forward (Bressler et al. 2004; Kim et al. 2007). This lack of selectivity of DMT1 is a major aspect of cadmium toxicity that has been reported elsewhere (Mackenzie et al. 2007; Kim et al. 2007) and will not be further discussed here.
Partly because DMT1 efficiently operates at low pH, most cells acquiring iron from the circulation rely on transferrin (Tf) and interaction with its receptor for their iron supply. The complex is internalized by endocytosis and iron is liberated at low pH in endosomes. The oxidation level of Tf-bound iron being ferric, the highly charged (trivalent) metal ion has to cross the endosomal membrane to reach the cytosol. From presently gathered evidence, ferric iron is first reduced in endosomes (Scheiber and Goldenberg 1993), probably by the ferric reductase Steap3 (Ohgami et al. 2005). Thus, ferrous iron crosses the endosomal membrane, again via DMT1 (Fleming et al. 1998), the same transporter involved in iron absorption by the body. The endosomal DMT1 form may be translated from an alternatively spliced transcript (Hubert and Hentze 2002), but none of the different DMT1 isoforms show strong divalent metal specificity in reconstituted systems (Mackenzie et al. 2007). Therefore DMT1 may also transport internalized Cd2+ from endosomes (Abouhamed et al. 2007) as it does for external cations. More recently, an alternative mediator of ferrous iron release from endosomes and lysosomes has been proposed (Dong et al. 2008). The TRPML1 (mucolipin 1, also known as MCOLN1) channel that is mutated in one type (IV) of mucolipidosis was associated with iron transport, and a large current was also measured with ferrous iron in TRPML2 transfected cells (Dong et al. 2008). Because these channels are not very selective among divalent cations, they may well help Cd2+ permeation through endosomal/lysosomal membranes, but this has not yet been addressed.
Distribution of cadmium in the body relies on its binding to carrier molecules in the circulation. Such molecules are proteins (Nordberg 2009), and serum Tf has been shown to bind Cd2+ (Harris and Madsen 1988). However, considering the apparent binding constant for Cd2+ compared to Fe3+, the proportion of Cd2+-bound Tf is probably very small, and the ability of the Tf receptor to bind and internalize Cd2+ loaded Tf has not been reported. The same is true for another Tf receptor, cubilin, that is most probably involved in the catabolism of the protein (Kozyraki et al. 2001). Cd2+ is also bound to other proteins than Tf in the circulation (Scott and Bradwell 1983). Among them, Cd2+ loaded MT is released by the liver and it is internalized also by cubilin in cells of the kidney proximal tubules (Abouhamed et al. 2007; Nordberg et al. 2009). This pathway contributes to accumulation of cadmium in the kidneys, but the interaction with iron homeostasis via this pathway is not documented.
The few other identified intracellular iron transporters have not been reported to be involved in Cd2+ trafficking. Surprisingly, the Cd2+ transport properties of the only well characterized iron exporter, ferroportin (FPN or MTP1, Ireg1, SLC40A1), are not yet known. This membrane protein releases ferrous iron into the circulation, from enterocytes, macrophages, and other cells, and it has a key regulatory role in systemic iron homeostasis (Ganz and Nemeth 2006). The involvement of FPN in Cd2+ distribution can now be probed with available tools (Rice et al. 2009), and it is of note that upregulation of the transporter upon Cd2+ exposure has been evidenced in enterocytes, macrophages, neurons, and other cells, e.g. (Min et al. 2008).
Iron homeostasis: regulation and cadmium interference
The nutritional iron status has a clear effect on Cd2+ absorption (Åkesson et al. 2002; Ryu et al. 2004). It follows that the mechanisms of cadmium toxicity must be considered with reference to the systems regulating different aspects of iron turnover in the body. Two specific systems are dedicated to iron homeostasis.
Systemic iron regulation is commonly associated with the synthesis and action of hepcidin (Hepc). This small hormone is mainly produced in the liver where iron needs and availability are detected, and it targets FPN for degradation at the basolateral membrane of enterocytes in particular (Ganz and Nemeth 2006). Therefore, Hepc represses iron absorption. Regulation of Hepc does not only depend on the iron status, as modified by anemia or hypoxia, but also on a number of other signals, such as inflammatory cytokines, with complex mechanisms that are being elucidated (Knutson 2009; Lee and Beutler 2009). Regarding the biochemistry of cadmium toxicity, Hepc may appear as an easy target of Cd2+ as cysteines make one-fourth of the mature Hepc amino acid sequence. However, no direct binding of Cd2+ to Hepc has been reported (Tselepis et al. 2010), and the effects of cadmium on fish Hepc synthesis and function (Chen et al. 2008) need to be extended to mammals and rationalized. As Hepc is a secreted hormone, its cysteines are involved in disulfide bridges, but the dynamics of this compact structure (Jordan et al. 2009) may leave room for coordination of cations under specific conditions (Tselepis et al. 2010). Also, precursor intracellular Hepc is likely to display free cysteines that may be reactive with any labile Cd2+ present inside Hepc-synthesizing cells. It will be necessary to discriminate between potential direct binding of Cd2+ to Hepc and mediated expression of cadmium intoxication on Hepc production to fully evaluate the impact of cadmium on systemic iron homeostasis.
Another level of regulation occurs in mammalian cells managing their individual iron supply and use. Cellular iron sensing is organized around cytosolic proteins, called Iron Regulatory Proteins (IRP) (Cairo and Recalcati 2007; Muckenthaler et al. 2008). Iron depletion or other stimuli, such as nitric oxide or hydrogen peroxide, activate IRP binding to regulated mRNA. Cadmium interacts with NO and redox signaling (Thévenod 2009), but Cd2+ and Zn2+ have also been shown to decrease IRP1 solubility (Martelli and Moulis 2004). Whereas the patho-physiological consequences of the latter observation remain to be fully explored, the cellular buildup of large Cd2+ concentrations, as observed in kidney cells of the proximal tubules for instance (Järup and Åkesson 2009), increases the turnover of IRP1 (Rousselet and Moulis 2008). In addition, the impact of cadmium on signaling pathways (see below) might also provide another way to influence the activity of Iron Regulatory Proteins, as they are substrates of kinases (Fillebeen et al. 2005; Wallander et al. 2006).
Iron use and cadmium interference
Cellular iron use is another process that may be perturbed by cadmium. Biosynthesis of iron-sulfur clusters mainly occurs in mitochondria, and it requires a machinery involving molecules containing reactive thiols, such as glutathione and glutaredoxin 5 (Rouault and Tong 2008; Lill 2009). It may thus be conceived that Cd2+ binds to some of these molecules and impairs the range of activities depending on iron-sulfur cofactors, especially cytosolic ones including IRP1 described above. The other biosynthetic pathway using large amounts of iron is heme production, predominantly for hemoglobinization of erythroblasts. Iron insertion into protoporphyrin IX (PPIX) is catalyzed by ferrochelatase in the mitochondrial matrix and Cd2+ inhibits this reaction (Fadigan and Dailey 1987). It should be noticed that iron deficiency is associated with ZnPPIX release, a marker of iron deficiency anemia (Labbé et al. 1999). Occurrence of CdPPIX has not been detected, but exposure of humans to a cadmium contaminated environment has been associated with increased, non iron-containing though metallated, PPIX derivatives (Staessen et al. 1996). Another step of heme biosynthesis may be sensitive to cadmium: δ-aminolevulinic acid dehydratase is a zinc-containing enzyme in mammals (Shoolingin-Jordan et al. 2002) which experiences changes of its kinetic properties in the presence of Cd2+ (Sommer and Beyersmann 1984).
Whereas the evidence for interference between cadmium exposure and either iron-sulfur or heme biosynthetic pathways is relatively scarce or indirect, other steps in heme turnover are sensitive to the toxic metal. Human erythrocytes experience increased apoptosis (eryptosis) when they are exposed to μM concentrations of CdCl2 for 2 days (Sopjani et al. 2008). From the two heme oxygenase isoforms, ubiquitous isoform 1 (HO-1) is strongly inducible at the transcriptional level by a large range of signals (Syapin 2008), including CdCl2 which enhances HO-1 transcription in HeLa cells via a short upstream cadmium response element (Takeda et al. 1994). The finding that several proteins bind to the cadmium response element (Sikorski et al. 2006; Koizumi et al. 2007) leaves open the question of how activation occurs, by the direct binding of cadmium to some transcription factor or by the stress enhancement of heat shock factor 1 or other transcriptional enhancer. In any case, the increase of HO activity induced by cadmium may contribute to signs of heme shortage, including anemia, observed upon cadmium insult, and it is a significant part of the cellular response to this oxidative stress.
More generally, the change in the cellular redox balance occurring in cells accumulating cadmium is likely to deregulate iron control by these cells. In contrast to zinc leakage, release of ferrous or ferric iron from metalloproteins is a major event maintaining and enhancing oxidative stress, for instance by conversion of oxygen derivatives into highly reactive hydroxyl radicals. Ferritin (FT) is the scavenger of liberated, otherwise uncontrolled, iron ions, and the strong expression of the FT genes under stress is an important cellular defense mechanism in a variety of conditions. Different binding sites for Cd2+ have been located in the mammalian FT structure (Granier et al. 1998), and some evidence for ex vivo cadmium binding to FT has been obtained from iron replete animals (Huebers et al. 1987). Cadmium-loaded sites may affect the exchange properties of FT with iron ions. Iron loaded FT is degraded by lysosomes and the proteasome with iron fluxes between vesicles and the cytosol (De Domenico et al. 2009), and lysosomes accumulate hemosiderin under conditions of iron overload. Whether FT-bound Cd2+ interferes with the cellular dynamics of FT-derived iron has not been reported, but cadmium association with this storage protein may help translocate the toxic metal. The role of FT in Cd2+ distribution remains to be addressed.
The variety of biological roles for iron is matched by the number of steps of iron homeostasis which can be perturbed by the presence of cadmium. From the preceding paragraphs, the experimental evidence for some possibilities (e.g. Cd2+ transport by DMT1, HO upregulation) is stronger than for others. But it should be clear from the above that the often proposed Fe/Cd metal substitution at the active sites of iron enzymes is not easy (see above Replacement of biological metals by cadmium) and it does not explain most aspects of cadmium perturbation of iron homeostasis.
Cadmium toxicity and manganese homeostasis
All other essential transition metals than zinc and iron occur at lower concentrations in Biology. The reason is probably that these other metals are mainly needed at the active sites of selected enzymes, whereas zinc and iron play additional roles, signaling for zinc, massive oxygen transport for iron, which require far larger amounts.
Very important biological reactions are catalyzed by manganese-containing enzymes. They mainly fall in four groups involved (i) in mitochondrial antioxidant defense via Mn-superoxide dismutase (Mn-SOD), (ii) in processing of nucleic acids with a range of nucleases, transposases and other enzymes having nucleic acids as substrates, (iii) in post-translational protein modifications with glycosyl- and sulfo-transferases, and (iv) in the conversion of glutamate and ammonia into glutamine by glutamine synthase. As this list shows, these activities occur in different cellular compartments, implying several transporters in different membranes permitting efficient trafficking of the metal.
Of note, it is still impossible today to assign an exclusive manganese transport activity to any membrane protein (Roth 2006), and the molecular pathways of manganese homeostasis are not fully identified in mammalian cells. Several oxidation states of manganese are accessible under physiological conditions. It thus can be proposed that the uptake mechanism by endocytosis of the Tf–Tf receptor complex allows manganese uptake as indicated for iron in Fig. 3. However, the experimental evidence for Mn3+ entry into cells via this route is contradictory, and it may not be active in many cases. For instance the role of Mn2+ oxidation and that of ceruloplasmin in manganese distribution may not be as simple as described above for iron (Jursa and Smith 2009). Furthermore, impairment of Tf endocytosis does not seem to be an important step in cadmium toxicology (see above Cadmium traffic and iron transport), and manganese/cadmium interference should not be looked for in this direction. Another less contradictory pathway for manganese entry into cells is via DMT1, the proton symport first identified for iron import (Gunshin et al. 1997) (see Cadmium traffic and iron transport). The involvement of iron transporters in manganese trafficking would imply that cadmium competes with both essential metals, to cross membranes at least.
But various observations in different cell types or animal models indicate that DMT1 cannot be the only manganese transporter (Erikson et al. 2007). Assuming that serum manganese is associated with organic anions (citrate, amino acids…) available in blood and other body fluids, manganese uptake may be mediated by organic anion transporters. But no tight binding chelators for manganese in the circulation have been identified with the exception of Tf for the Mn3+ oxidation state (Jursa and Smith 2009). Therefore, ionic manganese species may be substrates for other transporters (Roth 2006), and the ability of calcium channels to mediate manganese traffic is supported by pharmacological inhibition studies in some cell types (Heilig et al. 2006). Careful examination of cadmium sensitive tissues also identified transporters of the ZIP family (see above Molecular components involved in zinc trafficking) as mediators of cadmium uptake. Various levels of experimental evidence have been obtained for ZIP7 (Eide 2004), ZIP8, and ZIP14 (He et al. 2009; Himeno et al. 2009) involvement in cadmium transport. Cells derived from MT-deficient mice revealed that manganese and cadmium were competing for cellular uptake (Himeno et al. 2002), and further reports confirmed competition between Mn2+ and Cd2+ transport (Rousselet et al. 2008b; Himeno et al. 2009). A co-transport of Cd2+ and bicarbonate anion by ZIP8 is likely (He et al. 2009). However, Cd2+ uptake is inhibited by Zn2+ and other metal cations in Xenopus oocytes injected with ZIP8 cRNA, but not by Mn2+ or Fe2+ (Liu et al. 2008). ZIP8 may not be the only member of the family transporting Cd2+ since ZIP14 overproducing fibroblasts experience increased uptake of the toxic cation and of Mn2+ (Girijashanker et al. 2008; He et al. 2009). A possible difference between ZIP8 and ZIP14 is that the latter also transports Fe2+ (Lichten and Cousins 2009). It thus seems likely that Cd2+ toxicity can target manganese homeostasis at the level of ZIP transporters, but details and conditions under which this occurs have not yet been fully explored.
The initial characterization of ZIP8 in mice inbred strains and in adapted MT-null cells indicated large mRNA variations that were correlated with transport activity (Dalton et al. 2005; He et al. 2009; Fujishiro et al. 2009). However, the transporter responsible for Cd2+ uptake under different conditions and in different cells, whether ZIP8 or another molecule, is probably not simply regulated at the transcriptional level (Rousselet et al. 2008b; Satarug et al. 2008; Martin and Pognonec 2010). In the case of manganese, there is no known ‘manganese sensor’ in mammalian cells, as there are in bacteria (Papp-Wallace and Maguire 2006) or for iron in mammalian cells with IRP (see above Iron homeostasis: regulation and cadmium interference). It is thus not yet possible to propose that cadmium interference with manganese homeostasis concerns a specific manganese regulator in mammalian cells.
As stated above, once inside cells, manganese has also to reach different compartments. Candidate transporters for manganese loading into the Golgi apparatus belong to the secretory-pathway Ca2+-ATPases family. Despite their names, these ATP hydrolysis pumps of the P1B-type may have other physiological substrates than calcium, including Mn2+ (Vangheluwe et al. 2009) and cadmium. Although in vitro studies have addressed the effects of divalent metal ions on transferase activities found in the Golgi apparatus, the toxicological importance of manganese replacement by cadmium has not been demonstrated in vivo. The observation that cadmium intoxication impairs the proper folding of proteins (Kitamura and Hiramatsu 2010), including membrane and secreted ones, suggests that the presence of Cd2+ in the Golgi apparatus may inhibit some of these transferase activities.
It thus appears that different steps of manganese trafficking are sensitive to cadmium, and that manganese transporters seem privileged mediators of cadmium movements through biological membranes. However, it is not yet known whether cadmium toxicity is due to direct metal substitution at the active site of manganese-binding enzymes in mammalian cells, or to less direct mechanisms evoked by cadmium exposure and targeting manganese enzymes.
Cadmium toxicity, copper and other transition metals
Another transition metal of major importance in mammalian metabolism is copper. Very much like manganese, copper is involved at the active site of several enzymes, in most of which the electron exchange properties of copper (Cu+/2+) provide oxidoreductase activities. Examples are the largely distributed Cu–ZnSOD (Fig. 1), complex IV of the mitochondrial respiratory chain, multi-copper oxidases, such as ceruloplasmin and tyrosinase, and the secreted lysyloxidase. Again, as for manganese, distribution of copper must occur between different cellular compartments (Fig. 4).

Schematic representation of copper homeostasis relevant to cadmium toxicity. The symbols and cautious note of the legend of Fig. 2 are also valid for this figure. Abbreviations not defined in the text: APP, amyloid precursor protein; PrP, prion protein
Cellular copper import is mainly carried out by specific transporters of the Ctr family (SLC31) at the plasma membrane (Petris 2004). Reciprocally, the P1B-ATPases ATP7A and ATP7B, that are deficient in Menkes and Wilson diseases, respectively, are involved in cellular copper efflux from the cytosol. All these transporters are post-transcriptionally regulated as a function of intracellular copper concentration (van den Berghe and Klomp 2010). Pumps of the ATP7 family do not seem to efficiently transport cadmium, from the liver to the bile at least (Dijkstra et al. 1996), maybe because intracellular copper is mainly cuprous (1+) whereas cadmium is a divalent cation. Interestingly, although cadmium transport by P1B-ATPases is frequent throughout evolution up to plants (Morel et al. 2009), cation selectivity for some of them does occur (Lewinson et al. 2009). Exposure of animal cells to cadmium modifies the details of copper homeostasis, including for the above transporters (Chou et al. 2007), without any clear evidence for cadmium traffic via copper transporters in mammalian cells.
A likely point of convergence between cadmium toxicity and copper homeostasis is at the level of MT, the small protein that plays a central role in zinc homeostasis (see above Intracellular handling of zinc). Copper binding to thionein is even tighter than that of zinc or cadmium (Kägi and Schäffer 1988), and a role for copper-thionein in copper sensing coupled to metal-responsive transcription factor 1 (Chen et al. 2008) (see above Intracellular handling of zinc) may be proposed. The potential cadmium toxicity mechanism discussed above at this level for zinc may also apply to copper. However, an additional complexity of eukaryotic copper homeostasis is with the specialized proteins (the ‘copper chaperones’) that ferry copper to the enzymes needing it: examples are Atox1, Ccs, and Cox17 for ATP7A and B, Cu–ZnSOD, and cytochrome c oxidase, respectively, with intermediate proteins in the latter case (Kim et al. 2008). It follows that a set of high-affinity and selective proteins can compete for copper entering cells or copper released into the cytosol. Although MT on the one hand and chaperones on the other hand do not bind copper in the same redox state (cupric and cuprous, respectively), it is difficult to assign a copper-sensing function to any single protein. This statement includes Copper metabolism MURR1 domain containing (COMMD) or X-linked inhibitor of apoptosis (XIAP), proteins that may fulfill a similar sensing role (Maine and Burstein 2007; Mufti et al. 2007), and other proteins, such as the Amyloid Precursor Protein or the Prion Protein, the roles of which in copper homeostasis remain unclear. This complexity of copper regulation with proteins of seemingly overlapping functions does not help finding at which level cadmium may interrupt the finely tuned cellular use of copper.
However interaction between Cd2+ and copper chaperones cannot be dismissed (Takenouchi et al. 1999; Wernimont et al. 2000). Furthermore, cadmium transcriptionally upregulates the Prion Protein, as copper does, in rat PC12 pheochromocytoma cells (Varela-Nallar et al. 2006), and the toxic cation interferes with APP processing in the same and other cell lines (Smedman et al. 1997).
Therefore, the perturbation of copper homeostasis by cadmium has received experimental support, but the exact steps that are most sensitive to Cd2+ and that may determine the cellular response to the presence of the toxic cation are not clearly established.
Other essential transition metals, such as cobalt and molybdenum, participate in cellular metabolism but their concentrations are low in mammalian cells. Consequently perturbation of their homeostasis by cadmium is difficult to demonstrate. Vitamin B12 deficiency after cadmium accumulation cannot be ruled out (Frank et al. 2004), and some protection from cadmium toxicity may be afforded to rats by supplementation of cyanocobalamine (Couce et al. 1991). Also, mammalian molybdoenzymes can be inactivated by Cd2+ (Neumann and Leimkühler 2008), but such reactions have yet to be observed in vivo.
Cadmium toxicity and non-transition metals
For the sake of completeness, it must be clearly stated here that cadmium toxicity mechanisms do not only concern pathways that are responsible for the cellular control of transition metals. The other most significant essential metal cations in Biology are sodium, potassium, magnesium, and calcium. Cadmium has often been used as an in vitro pharmacological agent to study ion channels (Elinder and Arhem 2003), but the physiological relevance of the cadmium effects has virtually never received experimental support. This leaves open the possibility that the presence of Cd2+ modulates the activity and the transport properties of some of these channels in vivo (van Kerkhove and Swennen 2010). When compared with alkali and alkaline-earth metals, Cd2+ is most similar to Ca2+ with respect to size (see Replacement of biological metals by cadmium), implying that the desolvated ions can be competing substrates for calcium channels (Martelli et al. 2006).
But calcium transporters are not the only components of calcium homeostasis that are sensitive to cadmium. Several types of calcium-binding proteins, such as E–F hand proteins (calmodulin, troponin and others), γ-carboxyglutamic acid (Gla)-containing region of certain (prothrombin, factor IX…) vitamin K–dependent proteins, or calcium-dependent endonucleases, have been shown to bind Cd2+ at their calcium site (Fig. 1). Although it may be argued that most of these studies were carried out in vitro with purified proteins and with large cadmium concentrations, the occurrence of osteomalacia and osteoporosis upon human cadmium exposure (Bhattacharyya 2009) strongly suggests that several calcium dependent processes and proteins involved in bone resorption and formation are perturbed by the presence of cadmium. Beyond the osteotoxicity of cadmium, extracellular calcium proteins are more likely to suffer from cadmium insult than intracellular ones, as the toxic metal may be more readily available outside cells. In this respect, it is noteworthy that (E-) cadherin can bind Cd2+ with ensuing disruption of cell–cell junctions (Prozialeck and Edwards 2007). The ligands of Ca2+ in cadherins are mainly carboxylates (aspartates and glutamates) and a small number of amides (asparagines and glutamines) providing together a fully oxygenated coordination sphere for the cations. The dissociation constants for Ca2+ are a few tens of μM, suggesting a relative ease of substitution if the calcium concentration is not saturating. Indeed, examples of such substitutions in vitro have been shown (Fig. 1). Therefore, the direct cadmium binding to cadherins is probably among the most convincing examples of metal replacement as a toxicity mechanism for cadmium in vivo (Prozialeck and Edwards 2007). However, even in the case of adhesion molecules, it is not the only one at work to explain the effects of cadmium on mammalian cells. Recently reviewed experimental evidence (Prozialeck and Edwards 2007; Lee and Thévenod 2008) indicates that many calcium dependent pathways and proteins are sensitive to cadmium in an indirect way, with involvement of signaling cascades and other regulatory mechanisms.
Cadmium toxicity and pathways modulating the homeostasis of essential metals
In line with the conclusion of the preceding paragraph, it should be emphasized that many mechanisms of action of cadmium on mammalian cells are not directly interacting with pathways dedicated to the homeostasis of other metal cations. Cellular exposure to cadmium has been shown to change the status of a wide range of signaling systems. This topic has been thoroughly reviewed, including recently (Beyersmann and Hechtenberg 1997; Martelli et al. 2006; Thévenod 2009). A fast transient increase of intracellular Ca2+ concentration, with increased inositol-1,4,5 triphosphate, has been detected in different cell types exposed to low Cd2+ concentrations (sub-μM) before any transport may be significant. These features characterize ion-sensing G-protein coupled receptors that have been identified as calcium receptors (Chang and Shoback 2004). Cd2+ activates other receptors at the plasma membrane, such as the estrogens, the androgens, and other hormone receptors (Byrne et al. 2009; Iavicoli et al. 2009). Cd2+ binding to such receptors is more than three orders of magnitude tighter (dissociation constants below 1 nM) than binding to metalloproteins such as cadherins (dissociation constants in the μM range, see Cadmium toxicity and non-transition metals). Although details remain to be established (Höfer et al. 2009), the high affinity interaction of Cd2+ with surface receptors provides a tantalizing explanation to many cadmium effects, especially at low doses. Cd2+ also perturbs various intracellular signaling pathways including the different families of mitogenic protein kinases (MAPK), cAMP-dependent and calmodulin-dependent pathways (Thévenod 2009). Many of the cadmium-sensitive signaling pathways are zinc dependent (MAPK, PKC, etc.), most often with participation of zinc finger motifs. But lack of data in cellulo does not allow claiming that replacement of zinc by cadmium is responsible for the direct activation of these kinases. In any case, all the above signaling pathways are responsive to changing metal, including cadmium, concentrations with consequences on the homeostasis of essential metals. For instance, activation of MAPK and PKC induces MnSOD synthesis, and therefore manganese needs, in HepG2 hepatoma cells (Bianchi et al. 2002). Also, the activities of both IRP are modulated by phosphorylation events in which PKC may play a part (Fillebeen et al. 2005; Wallander et al. 2006).
The same phenomena just described for signaling pathways also pertain to transcription factors, such as NF-κB, products of the oncogenes c-jun, c-myc, egr-1 (Beyersmann and Hechtenberg 1997), and others (Nrf2, Sp1, CREB, etc.), most of them bearing zinc binding motifs (fingers). A list of genes sensitive to cadmium exposure in hepatocytes has been recently published (Hsiao and Stapleton 2009). Many of these transcription factors enhance the expression of genes encoding Cd2+ sensitive proteins (Murata et al. 1999). MT and HO have been presented above. The limiting enzyme of glutathione (GSH) biosynthesis, γ-glutamyl-cysteine synthase, and heat shock proteins belong to the same list. The corresponding activities contribute to intracellular cadmium management and to minimize impairment of cellular functions such as protein folding. Also, transcription of many genes encoding metalloproteins is under the dependence of cadmium-activated transcription factors. Examples are expression of the MnSOD gene by AP-1 (dimers of c-Jun, c-Fos and other proteins), and those of Cu-ZnSOD, HO-1, and FT by Nrf2. Consequently, the homeostasis of metals have to adjust to the transcriptional changes induced by cadmium.
Intracellular Cd2+ may bind to a variety of bio-molecules (see Replacement of biological metals by cadmium), especially when the binding capacities of MT and glutathione are exceeded. In particular the shielding of reactive cysteine thiolates is expected to interfere with proper building of disulfide bridges in proteins and with the cellular buffering potential against oxidative species. This is thought to be the main underlying mechanism for the pro-oxidant properties of cadmium, at least upon acute exposure (Liu et al. 2009; Cuypers et al. 2010). Under these conditions, enhanced production of partly reduced and highly reactive oxygen species (the reactive oxygen species: ROS) alters expression of genes under the dependence of redox-sensitive transcription factors (see above), impacts signal transduction pathways, and ROS, particularly at high concentrations, can liberate metal cations from proteins and enzymes. The released cations should be scavenged by metal-trapping molecules such as GSH, MT, FT, and a few others. But, under relentless ROS producing conditions, iron, copper, and manganese catalyze an even larger production of these species via the so-called Fenton reactions. Cadmium does not have to replace the endogenous metals at the active sites of enzymes to abolish biological activities, since ROS are the most convincing mediators of the above effects.
The mechanistic scenarios presented in the preceding paragraph may apply to other aspects of cadmium toxicity such as inhibition of DNA repair and perturbed protein processing. These topics are discussed in more detail elsewhere (Beyersmann and Hartwig 2008; Kitamura and Hiramatsu 2010; Liu et al. 2009; Hartwig 2010).
Cellular resistance to cadmium
Most mechanisms of cadmium toxicity studied in the laboratory mainly apply to the acute, and often heavy, exposure of naive mammalian cells to the toxic metal. However, it is likely that under chronic exposure additional modes of action are at play. Different types of cell death have been observed by exposing cells to cadmium, but long term contact may allow survival and even growth, often after a period of quiescence. For cells continuously exposed to cadmium, sustained upregulation of protection molecules has been observed (Hart et al. 2001), with adjustment of signaling pathways (Oh et al. 2009). Decreased apoptosis does not always correlate with increased production of anti-oxidant molecules. Overproduction of calbindin D28 in human U937 monocytes (Jeon et al. 2004), decreased phosphorylation of c-Jun N-terminal kinase in rat pulmonary epithelial cells (Lau et al. 2006), and Bcl-2 and Bax over and down-expression, respectively, in mouse testicular Leydig cells (Singh et al. 2009) all led to the same outcome of decreased apoptosis. MT depleted cells have provided useful models in which removal of the most immediate cellular response to cadmium revealed alternative pathways of resistance. Fibroblasts of MT−/− murine embryos adapted to cadmium evidenced inhibition of a T-type voltage-dependent cationic channel as a way of decreasing Cd2+ uptake (Leslie et al. 2006) and competition of Cd2+ import by Mn2+ addition (Himeno et al. 2002, 2009). Inhibition of ZIP8 and ZIP14 transcription was detected in such cells (Fujishiro et al. 2006, 2009).
A more realistic situation in a polluted environment than exposure to only cadmium is that of a combination of different compounds interacting with cells. For instance, human populations living in regions where zinc smelters are or were in use (Thomas et al. 2009), workers involved in treating spent nickel–cadmium batteries, or the general population in heavily industrialized areas (Järup 2003) experience concomitant exposure to several metals among many pollutants.
Therefore, instead of selecting cells having developed resistance against cadmium, it may be relevant to expose them to large concentrations of a more abundant and ‘safer’ metal that occurs with cadmium in the environment. Zinc in areas surrounding smelters is an example. HeLa cells were grown for several weeks in the presence of increasing zinc concentrations (Chimienti et al. 2001). The selected cellular population displayed accumulation of intracellular zinc and upregulation of MT, but also increased sensitivity to oxidative stress in the form of hydrogen peroxide (Chimienti et al. 2001). The resistance against high zinc concentrations developed by adapted HeLa cells imparts these cells with high resistance against cadmium salts (Rousselet et al. 2008b). But, instead of safely accumulating the metal as in the case of Zn2+, resistant cells have developed the ability to repress Cd2+ uptake upon long-term high zinc exposure. The transporter responsible for cadmium uptake appears to be the same as the one for manganese entry into HeLa cells (Rousselet et al. 2008b). This result is similar to the conclusion obtained with MT−/− fibroblasts (Himeno et al. 2002), but the regulatory level at which this transporter is inhibited seems different (Rousselet et al. 2008b; Fujishiro et al. 2006). In addition, a proteomic study evidenced an unexpected link between adaptation to high zinc concentrations and increased catabolism of tyrosine, a process which has a role in the sensitivity to cadmium (Rousselet et al. 2008a). This example shows that all details of the cellular response to cadmium must be further investigated. Although zinc often affords protection against cadmium toxicity, HeLa cells adapted to high zinc concentrations display changed cellular handling of cadmium, manganese, and calcium (Rousselet et al. 2008a, b). This cellular model also illustrates the conclusions of a review written more than 10 years ago (Beyersmann and Hechtenberg 1997) emphasizing the interference between cadmium toxicity and the homeostasis of essential metals, and indicating the gaps in the knowledge about the mechanisms of cadmium toxicity. The last decade has seen progress in further discovering the complexity of these mechanisms but the integration of all observations in a general and convincing model of cadmium toxicity is still awaited.
Open questions and outlook
Although many important industrial uses of cadmium have now been banned, the cleaning of the metal from polluted areas is difficult, and natural or anthropogenic dispersion of cadmium in the environment will continue in the years to come. Some of the unique properties of this metal will probably be hard to replace by safer substitutes for some present or future applications. Therefore, human contact with cadmium will not stop soon, and a deeper insight into the mechanisms of cadmium toxicity appears as the only way of designing proper protective actions for exposed populations. Beside the health problems generated by this ubiquitous pollutant, the range of bio-molecules targeted by cadmium raises valuable biological questions, some of which have been detailed above.
Focusing on biological cations, the main question is not whether cadmium impacts the homeostasis of metals, but how it does so and to what extent. Although there should be no doubt that Cd2+ can replace other cations at different sites in proteins and other biological molecules, the single explanation by substitution fails to explain the toxicological effects of cadmium, particularly at low dose. Indeed, in most instances, the exogenous pollutant cannot quantitatively compete with endogenous essential cations, especially calcium, zinc, iron, and even copper or manganese. Cadmium does accumulate mainly in specific organs of the body throughout life (Järup and Åkesson 2009; Nawrot et al. 2010; Nordberg et al. 2009; Satarug et al. 2010), but under which actual chemical form(s) remains unknown. Part of it is clearly associated with MT, some is likely bound to GSH, but such interactions are dynamic, both as for the binding of Cd2+ and the localization of the cadmium containing complexes. Furthermore, intracellular stores of cadmium (endoplasmic reticulum, acidic vesicles, mitochondria, nucleus) have been phenomenologically suggested, but their relative ease of mobilization has been rarely figured out. This question is key to understand biological Cd2+ effects, but efficient and direct tools are still lacking for such studies. Therefore, no strong biological evidence of metal substitution by Cd2+ affords a convincing conceptual frame to explain biological effects.
The impact of cadmium on the homeostasis of other metals reviewed here goes far beyond mere metal substitutions, and they include indirect effects mediated in part by redox sensitive transcription and transduction pathways. Transport (Fe2+ and DMT1, Mn2+ and ZIP8 …) and distribution (Zn2+ and thioneins) of endogenous metals (Figs. 2, 3, 4) provide efficient ways for Cd2+ traffic. Once Cd2+ is in the vicinity of cells or inside them a variety of biological interferences occur.
However, a detailed knowledge of these interferences is still limited by pending questions. Among them, the following, most closely related to the topics of this review, can be listed:
-
Which transporters allow Cd2+ to cross biological membranes, in which cells, and under which conditions? What determines the cation specificity of these transporters?
-
How (in what kind of molecular association) is Cd2+ accumulated inside cells? in which compartments? How does Cd2+ build-up changes the dynamics of exchange for other cations such as Ca2+, Zn2+, and Fe2+/3+?
-
Is Cd2+ targeting regulatory molecules dedicated to the homeostasis of specific metals, such as Hepc, FT, or IRP for iron, or chaperones for copper?
These questions, some of which are indicated in Figs. 2, 3, 4, arise in part from the still large gaps in the available knowledge in the homeostasis of essential metals. Topics such as zinc molecular sensing and distribution, copper regulation, manganese traffic, and many others are only emerging. Furthermore, which mechanism of cadmium toxicity predominates and which metal homeostatic and signaling pathways are impacted are likely to be cell specific and dependent on the detailed cellular environment. At the end of this review we must admit that the extent to which any process contributes to the overall cellular toxicity of cadmium is most often not known. It is certainly a major challenge of the years to come to frame cadmium, and other toxics, studies within a global description of cellular behavior. Then it must be hoped that the emerging methods of Systems Biology as applied to predictive toxicology will help reaching this goal.
Abbreviations
- DMT1:
-
Divalent metal-ion transporter 1 (SLC11A2)
- FPN:
-
Ferroportin (SLC40A1)
- FT:
-
Ferritin
- Hepc:
-
Hepcidin
- IRP:
-
Iron regulatory protein(s)
- MAPK:
-
Mitogen activated protein kinases
- MT:
-
Metallothionein(s)
- MTF-1:
-
Metal-response element-binding transcription factor
- PKC:
-
Protein kinase C
- PPIX:
-
Protoporphyrin IX
- ROS:
-
Reactive oxygen species
- HO-1:
-
Heme oxygenase 1
- SOD:
-
Superoxide dismutase
- Tf:
-
Transferrin
- ZnT:
-
Zinc transporter (SLC30)
- ZIP:
-
Zinc regulated and iron regulated metal transporter-like protein(s) (SLC39)
References
Abouhamed M, Wolff NA, Lee WK, Smith CP, Thévenod F (2007) Knockdown of endosomal/lysosomal divalent metal transporter 1 by RNA interference prevents cadmium-metallothionein-1 cytotoxicity in renal proximal tubule cells. Am J Physiol Renal Physiol 293:F705–F712
Åkesson A, Berglund M, Schütz A, Bjellerup P, Bremme K, Vahter M (2002) Cadmium exposure in pregnancy and lactation in relation to iron status. Am J Public Health 92:284–287
Auld DS (2001) Zinc coordination sphere in biochemical zinc sites. Biometals 14:271–313
Berg JM, Godwin HA (1997) Lessons from zinc-binding peptides. Annu Rev Biophys Biomol Struct 26:357–371
Beyersmann D, Haase H (2001) Functions of zinc in signaling, proliferation and differentiation of mammalian cells. Biometals 14:331–341
Beyersmann D, Hartwig A (2008) Carcinogenic metal compounds: recent insight into molecular and cellular mechanisms. Arch Toxicol 82:493–512
Beyersmann D, Hechtenberg S (1997) Cadmium, gene regulation, and cellular signalling in mammalian cells. Toxicol Appl Pharmacol 144:247–261
Bhattacharyya MH (2009) Cadmium osteotoxicity in experimental animals: mechanisms and relationship to human exposures. Toxicol Appl Pharmacol 238:258–265
Bianchi A, Becuwe P, Franck P, Dauca M (2002) Induction of MnSOD gene by arachidonic acid is mediated by reactive oxygen species and p38 MAPK signaling pathway in human HepG2 hepatoma cells. Free Radic Biol Med 32:1132–1142
Bressler JP, Olivi L, Cheong JH, Kim Y, Bannon D (2004) Divalent metal transporter 1 in lead and cadmium transport. Ann N Y Acad Sci 1012:142–152
Byrne C, Divekar SD, Storchan GB, Parodi DA, Martin MB (2009) Cadmium—a metallohormone? Toxicol Appl Pharmacol 238:266–271
Cairo G, Recalcati S (2007) Iron-regulatory proteins: molecular biology and pathophysiological implications. Expert Rev Mol Med 9:1–13
Carvalho E, Gothe PO, Bauer R, Danielsen E, Hemmingsen L (1995) Effect of inhibitors on the coordination geometries of cadmium at the metal sites in angiotensin-I-converting enzyme. Eur J Biochem 234:780–785
Chang W, Shoback D (2004) Extracellular Ca2+-sensing receptors—an overview. Cell Calcium 35:183–196
Chen J, Shi YH, Li MY (2008a) Changes in transferrin and hepcidin genes expression in the liver of the fish Pseudosciaena crocea following exposure to cadmium. Arch Toxicol 82:525–530
Chen X, Hua H, Balamurugan K, Kong X, Zhang L, George GN, Georgiev O, Schaffner W, Giedroc DP (2008b) Copper sensing function of Drosophila metal-responsive transcription factor-1 is mediated by a tetranuclear Cu(I) cluster. Nucleic Acids Res 36:3128–3138
Chimienti F, Jourdan E, Favier A, Sève M (2001) Zinc resistance impairs sensitivity to oxidative stress in HeLa cells: protection through metallothioneins expression. Free Radic Biol Med 31:1179–1190
Chou DK, Zhao Y, Gao S, Chou IN, Toselli P, Stone P, Li W (2007) Perturbation of copper (Cu) homeostasis and expression of Cu-binding proteins in cadmium-resistant lung fibroblasts. Toxicol Sci 99:267–276
Couce MD, Varela JM, Sanchez A, Casas JS, Sordo J, Lopez-Rivadulla M (1991) Effects of vitamin B12 on cadmium toxicity in rats. J Inorg Biochem 41:1–6
Cuypers A, Plusquin M, Remans T, Jozefczak M, Keunen E, Gielen H, Opdenakker K, Nair AR, Munters E, Nawrot T, Vangronsveld J, Smeets K (2010) Cadmium: an oxidative challenge. Biometals. doi:10.1007/s10534-010-9329-x
Dalton TP, He L, Wang B, Miller ML, Jin L, Stringer KF, Chang X, Baxter CS, Nebert DW (2005) Identification of mouse SLC39A8 as the transporter responsible for cadmium-induced toxicity in the testis. Proc Natl Acad Sci USA 102:3401–3406
De Domenico I, Ward DM, Kaplan J (2009) Specific iron chelators determine the route of ferritin degradation. Blood 114:4546–4551
Dijkstra M, Havinga R, Vonk RJ, Kuipers F (1996) Bile secretion of cadmium, silver, zinc and copper in the rat. Involvement of various transport systems. Life Sci 59:1237–1246
Dong XP, Cheng X, Mills E, Delling M, Wang F, Kurz T, Xu H (2008) The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 455:992–996
Edwards JR, Prozialeck WC (2009) Cadmium, diabetes and chronic kidney disease. Toxicol Appl Pharmacol 238:289–293
Eide DJ (2004) The SLC39 family of metal ion transporters. Pflugers Arch 447:796–800
El Heni J, Messaoudi I, Hammouda F, Kerkeni A (2009) Protective effects of selenium (Se) and zinc (Zn) on cadmium (Cd) toxicity in the liver of the rat: effects on the oxidative stress. Ecotoxicol Environ Saf 72:1559–1564
Elinder F, Arhem P (2003) Metal ion effects on ion channel gating. Q Rev Biophys 36:373–427
Eng BH, Guerinot ML, Eide D, Saier MH Jr (1998) Sequence analyses and phylogenetic characterization of the ZIP family of metal ion transport proteins. J Membr Biol 166:1–7
Erikson KM, Thompson K, Aschner J, Aschner M (2007) Manganese neurotoxicity: a focus on the neonate. Pharmacol Ther 113:369–377
Fadigan A, Dailey HA (1987) Inhibition of ferrochelatase during differentiation of murine erythroleukaemia cells. Biochem J 243:419–424
Fillebeen C, Caltagirone A, Martelli A, Moulis J-M, Pantopoulos K (2005) IRP1 Ser-711 is a phosphorylation site, critical for regulation of RNA-binding and aconitase activities. Biochem J 388:143–150
Fleming MD, Romano MA, Su MA, Garrick LM, Garrick MD, Andrews NC (1998) Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc Natl Acad Sci USA 95:1148–1153
Frank A, McPartlin J, Danielsson R (2004) Nova Scotia moose mystery—a moose sickness related to cobalt- and vitamin B12 deficiency. Sci Total Environ 318:89–100
Frederickson CJ, Bush AI (2001) Synaptically released zinc: physiological functions and pathological effects. Biometals 14:353–366
Frederickson CJ, Koh JY, Bush AI (2005) The neurobiology of zinc in health and disease. Nat Rev Neurosci 6:449–462
Fried W (2009) Erythropoietin and erythropoiesis. Exp Hematol 37:1007–1015
Fujishiro H, Okugaki S, Nagao S, Satoh M, Himeno S (2006) Characterization of gene expression profiles of metallothionein-null cadmium-resistant cells. J Health Sci 52:292–299
Fujishiro H, Okugaki S, Yasumitsu S, Enomoto S, Himeno S (2009) Involvement of DNA hypermethylation in down-regulation of the zinc transporter ZIP8 in cadmium-resistant metallothionein-null cells. Toxicol Appl Pharmacol 241:195–201
Ganz T, Nemeth E (2006) Regulation of iron acquisition and iron distribution in mammals. Biochim Biophys Acta 1763:690–699
Garrick MD, Garrick LM (2009) Cellular iron transport. Biochim Biophys Acta 1790:309–325
Girijashanker K, He L, Soleimani M, Reed JM, Li H, Liu Z, Wang B, Dalton TP, Nebert DW (2008) Slc39a14 gene encodes ZIP14, a metal/bicarbonate symporter: similarities to the ZIP8 transporter. Mol Pharmacol 73:1413–1423
Granier T, Comberton G, Gallois B, d’Estaintot BL, Dautant A, Crichton RR, Precigoux G (1998) Evidence of new cadmium binding sites in recombinant horse L-chain ferritin by anomalous Fourier difference map calculation. Proteins 31:477–485
Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA (1997) Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388:482–488
Gyulkhandanyan AV, Lee SC, Bikopoulos G, Dai F, Wheeler MB (2006) The Zn-transporting pathways in pancreatic beta-cells: a role for the L-type voltage-gated Ca2+ channel. J Biol Chem 281:9361–9372
Haase H, Beyersmann D (2002) Intracellular zinc distribution and transport in C6 rat glioma cells. Biochem Biophys Res Commun 296:923–928
Haase H, Rink L (2009) Functional significance of zinc-related signaling pathways in immune cells. Annu Rev Nutr 29:133–152
Hamer DH (1986) Metallothionein. Annu Rev Biochem 55:913–951
Harris WR, Madsen LJ (1988) Equilibrium studies on the binding of cadmium(II) to human serum transferrin. Biochemistry 27:284–288
Hart BA, Potts RJ, Watkin RD (2001) Cadmium adaptation in the lung—a double-edged sword? Toxicology 160:65–70
Hartwig A (2010) Mechanisms in cadmium-induced carcinogenicity: recent insights. Biometals. doi:10.1007/s10534-010-9330-4
He L, Wang B, Hay EB, Nebert DW (2009) Discovery of ZIP transporters that participate in cadmium damage to testis and kidney. Toxicol Appl Pharmacol 238:250–257
Heilig EA, Thompson KJ, Molina RM, Ivanov AR, Brain JD, Wessling-Resnick M (2006) Manganese and iron transport across pulmonary epithelium. Am J Physiol Lung Cell Mol Physiol 290:L1247–L1259
Himeno S, Yanagiya T, Enomoto S, Kondo Y, Imura N (2002) Cellular cadmium uptake mediated by the transport system for manganese. Tohoku J Exp Med 196:43–50
Himeno S, Yanagiya T, Fujishiro H (2009) The role of zinc transporters in cadmium and manganese transport in mammalian cells. Biochimie 91:1218–1222
Höfer N, Diel P, Wittsiepe J, Wilhelm M, Degen GH (2009) Dose- and route-dependent hormonal activity of the metalloestrogen cadmium in the rat uterus. Toxicol Lett 191:123–131
Horiguchi H, Kayama F, Oguma E, Willmore WG, Hradecky P, Bunn HF (2000) Cadmium and platinum suppression of erythropoietin production in cell culture: clinical implications. Blood 96:3743–3747
Hsiao CJ, Stapleton SR (2009) Early sensing and gene expression profiling under a low dose of cadmium exposure. Biochimie 91:329–343
Hubert N, Hentze MW (2002) Previously uncharacterized isoforms of divalent metal transporter (DMT)-1: implications for regulation and cellular function. Proc Natl Acad Sci USA 99:12345–12350
Huebers HA, Huebers E, Csiba E, Rummel W, Finch CA (1987) The cadmium effect on iron absorption. Am J Clin Nutr 45:1007–1012
Iavicoli I, Fontana L, Bergamaschi A (2009) The effects of metals as endocrine disruptors. J Toxicol Environ Health B Crit Rev 12:206–223
Jacquillet G, Barbier O, Cougnon M, Tauc M, Namorado MC, Martin D, Reyes JL, Poujeol P (2006) Zinc protects renal function during cadmium intoxication in the rat. Am J Physiol Renal Physiol 290:F127–F137
Jansen J, Karges W, Rink L (2009) Zinc and diabetes—clinical links and molecular mechanisms. J Nutr Biochem 20:399–417
Järup L (2003) Hazards of heavy metal contamination. Br Med Bull 68:167–182
Järup L, Åkesson A (2009) Current status of cadmium as an environmental health problem. Toxicol Appl Pharmacol 238:201–208
Jeon HK, Jin HS, Lee DH, Choi WS, Moon CK, Oh YJ, Lee TH (2004) Proteome analysis associated with cadmium adaptation in U937 cells: identification of calbindin-D28k as a secondary cadmium-responsive protein that confers resistance to cadmium-induced apoptosis. J Biol Chem 279:31575–31583
Johri N, Jacquillet G, Unwin R (2010) Heavy metal poisoning: the effects of cadmium on the kidney. BioMetals. doi:10.1007/s10534-010-9328-y
Jordan JB, Poppe L, Haniu M, Arvedson T, Syed R, Li V, Kohno H, Kim H, Schnier PD, Harvey TS, Miranda LP, Cheetham J, Sasu BJ (2009) Hepcidin revisited, disulfide connectivity, dynamics, and structure. J Biol Chem 284:24155–24167
Jursa T, Smith DR (2009) Ceruloplasmin alters the tissue disposition and neurotoxicity of manganese, but not its loading onto transferrin. Toxicol Sci 107:182–193
Kägi JH, Schäffer A (1988) Biochemistry of metallothionein. Biochemistry 27:8509–8515
Kim DW, Kim KY, Choi BS, Youn P, Ryu DY, Klaassen CD, Park JD (2007) Regulation of metal transporters by dietary iron, and the relationship between body iron levels and cadmium uptake. Arch Toxicol 81:327–334
Kim BE, Nevitt T, Thiele DJ (2008) Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol 4:176–185
Kippler M, Goessler W, Nermell B, Ekström EC, Lönnerdal B, El Arifeen S, Vahter M (2009) Factors influencing intestinal cadmium uptake in pregnant Bangladeshi women—a prospective cohort study. Environ Res 109:914–921
Kitamura M, Hiramatsu N (2010) The oxidative stress—endoplasmic reticulum stress axis in cadmium toxicity. Biometals. doi:10.1007/s10534-010-9296-2
Knutson MD (2009) Into the matrix: regulation of the iron regulatory hormone hepcidin by matriptase-2. Nutr Rev 67:284–288
Koizumi S, Gong P, Suzuki K, Murata M (2007) Cadmium-responsive element of the human heme oxygenase-1 gene mediates heat shock factor 1-dependent transcriptional activation. J Biol Chem 282:8715–8723
Kozyraki R, Fyfe J, Verroust PJ, Jacobsen C, Dautry-Varsat A, Gburek J, Willnow TE, Christensen EI, Moestrup SK (2001) Megalin-dependent cubilin-mediated endocytosis is a major pathway for the apical uptake of transferrin in polarized epithelia. Proc Natl Acad Sci USA 98:12491–12496
Labbé RF, Vreman HJ, Stevenson DK (1999) Zinc protoporphyrin: a metabolite with a mission. Clin Chem 45:2060–2072
Laity JH, Andrews GK (2007) Understanding the mechanisms of zinc-sensing by metal-response element binding transcription factor-1 (MTF-1). Arch Biochem Biophys 463:201–210
Lane TW, Morel FM (2000) A biological function for cadmium in marine diatoms. Proc Natl Acad Sci USA 97:4627–4631
Lau AT, Zhang J, Chiu JF (2006) Acquired tolerance in cadmium-adapted lung epithelial cells: roles of the c-Jun N-terminal kinase signaling pathway and basal level of metallothionein. Toxicol Appl Pharmacol 215:1–8
Lee PL, Beutler E (2009) Regulation of hepcidin and iron-overload disease. Annu Rev Pathol 4:489–515
Lee WK, Thévenod F (2008) Novel roles for ceramides, calpains and caspases in kidney proximal tubule cell apoptosis: lessons from in vitro cadmium toxicity studies. Biochem Pharmacol 76:1323–1332
Leslie EM, Liu J, Klaassen CD, Waalkes MP (2006) Acquired cadmium resistance in metallothionein-I/II(−/−) knockout cells: role of the T-type calcium channel Cacnalpha1G in cadmium uptake. Mol Pharmacol 69:629–639
Lewinson O, Lee AT, Rees DC (2009) A P-type ATPase importer that discriminates between essential and toxic transition metals. Proc Natl Acad Sci USA 106:4677–4682
Lichten LA, Cousins RJ (2009) Mammalian zinc transporters: nutritional and physiologic regulation. Annu Rev Nutr 29:153–176
Lichtlen P, Wang Y, Belser T, Georgiev O, Certa U, Sack R, Schaffner W (2001) Target gene search for the metal-responsive transcription factor MTF-1. Nucleic Acids Res 29:1514–1523
Lill R (2009) Function and biogenesis of iron–sulphur proteins. Nature 460:831–838
Lindert U, Cramer M, Meuli M, Georgiev O, Schaffner W (2009) Metal-responsive transcription factor 1 (MTF-1) activity is regulated by a nonconventional nuclear localization signal and a metal-responsive transactivation domain. Mol Cell Biol 29:6283–6293
Liu Z, Li H, Soleimani M, Girijashanker K, Reed JM, He L, Dalton TP, Nebert DW (2008) Cd2+ versus Zn2+ uptake by the ZIP8 HCO3-dependent symporter: kinetics, electrogenicity and trafficking. Biochem Biophys Res Commun 365:814–820
Liu J, Qu W, Kadiiska MB (2009) Role of oxidative stress in cadmium toxicity and carcinogenesis. Toxicol Appl Pharmacol 238:209–214
Mackenzie B, Takanaga H, Hubert N, Rolfs A, Hediger MA (2007) Functional properties of multiple isoforms of human divalent metal-ion transporter 1 (DMT1). Biochem J 403:59–69
Maine GN, Burstein E (2007) COMMD proteins: COMMing to the scene. Cell Mol Life Sci 64:1997–2005
Maret W (2008) Metallothionein redox biology in the cytoprotective and cytotoxic functions of zinc. Exp Gerontol 43:363–369
Maret W (2009) Molecular aspects of human cellular zinc homeostasis: redox control of zinc potentials and zinc signals. Biometals 22:149–157
Maret W, Krężel A (2007) Cellular zinc and redox buffering capacity of metallothionein/thionein in health and disease. Mol Med 13:371–375
Maret W, Li Y (2009) Coordination dynamics of zinc in proteins. Chem Rev 109:4682–4707
Margoshes M, Vallee BL (1957) A cadmium protein from equine kidney cortex. J Am Chem Soc 79:4813–4814
Martelli A, Moulis J-M (2004) Zinc and cadmium specifically interfere with RNA-binding activity of human iron regulatory protein 1. J Inorg Biochem 98:1413–1420
Martelli A, Rousselet E, Dycke C, Bouron A, Moulis J-M (2006) Cadmium toxicity in animal cells by interference with essential metals. Biochimie 88:1807–1814
Martin P, Pognonec P (2010) ERK and cell death: cadmium toxicity, sustained ERK activation and cell death. FEBS J 277:39–46
Matsuoka M, Igisu H (2001) Cadmium induces phosphorylation of p53 at serine 15 in MCF-7 cells. Biochem Biophys Res Commun 282:1120–1125
Méplan C, Mann K, Hainaut P (1999) Cadmium induces conformational modifications of wild-type p53 and suppresses p53 response to DNA damage in cultured cells. J Biol Chem 274:31663–31670
Méplan C, Richard MJ, Hainaut P (2000) Metalloregulation of the tumor suppressor protein p53: zinc mediates the renaturation of p53 after exposure to metal chelators in vitro and in intact cells. Oncogene 19:5227–5236
Min KS, Ueda H, Kihara T, Tanaka K (2008) Increased hepatic accumulation of ingested Cd is associated with upregulation of several intestinal transporters in mice fed diets deficient in essential metals. Toxicol Sci 106:284–289
Morel FM (2008) The co-evolution of phytoplankton and trace element cycles in the oceans. Geobiology 6:318–324
Morel M, Crouzet J, Gravot A, Auroy P, Leonhardt N, Vavasseur A, Richaud P (2009) AtHMA3, a P1B-ATPase allowing Cd/Zn/Co/Pb vacuolar storage in Arabidopsis. Plant Physiol 149:894–904
Muckenthaler MU, Galy B, Hentze MW (2008) Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu Rev Nutr 28:197–213
Mufti AR, Burstein E, Duckett CS (2007) XIAP: cell death regulation meets copper homeostasis. Arch Biochem Biophys 463:168–174
Murata M, Gong P, Suzuki K, Koizumi S (1999) Differential metal response and regulation of human heavy metal-inducible genes. J Cell Physiol 180:105–113
Nakazato K, Nagamine T, Suzuki K, Kusakabe T, Moon HD, Oikawa M, Sakai T, Arakawa K (2008) Subcellular changes of essential metal shown by in-air micro-PIXE in oral cadmium-exposed mice. Biometals 21:83–91
Nawrot TS, Staessen JA, Roels HA, Munters E, Cuypers A, Richart T, Ruttens A, Smeets K, Clijsters H, Vangronsveld J (2010) Cadmium exposure in the population: from health risks to strategies of prevention. BioMetals. doi:10.1007/s10534-010-9343-z
Neumann M, Leimkühler S (2008) Heavy metal ions inhibit molybdoenzyme activity by binding to the dithiolene moiety of molybdopterin in Escherichia coli. FEBS J 275:5678–5689
Nordberg GF (2009) Historical perspectives on cadmium toxicology. Toxicol Appl Pharmacol 238:192–200
Nordberg GF, Jin T, Wu X, Lu J, Chen L, Lei L, Hong F, Nordberg M (2009) Prevalence of kidney dysfunction in humans—relationship to cadmium dose, metallothionein, immunological and metabolic factors. Biochimie 91:1282–1285
Oh SH, Lee SY, Choi CH, Lee SH, Lim SC (2009) Cadmium adaptation is regulated by multidrug resistance-associated protein-mediated Akt pathway and metallothionein induction. Arch Pharm Res 32:883–891
Ohana E, Hoch E, Keasar C, Kambe T, Yifrach O, Hershfinkel M, Sekler I (2009) Identification of the Zn2+ binding site and mode of operation of a mammalian Zn2+ transporter. J Biol Chem 284:17677–17686
Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, Fleming MD (2005) Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet 37:1264–1269
Palmiter RD, Findley SD (1995) Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J 14:639–649
Pan J, Plant JA, Voulvoulis N, Oates CJ, Ihlenfeld C (2010) Cadmium levels in Europe: implications for human health. Environ Geochem Health 32:1–12
Papp-Wallace KM, Maguire ME (2006) Manganese transport and the role of manganese in virulence. Annu Rev Microbiol 60:187–209
Petris MJ (2004) The SLC31 (Ctr) copper transporter family. Pflugers Arch 447:752–755
Prozialeck WC, Edwards JR (2007) Cell adhesion molecules in chemically-induced renal injury. Pharmacol Ther 114:74–93
Rice AE, Mendez MJ, Hokanson CA, Rees DC, Bjorkman PJ (2009) Investigation of the biophysical and cell biological properties of ferroportin, a multipass integral membrane protein iron exporter. J Mol Biol 386:717–732
Rogalska J, Brzóska MM, Roszczenko A, Moniuszko-Jakoniuk J (2009) Enhanced zinc consumption prevents cadmium-induced alterations in lipid metabolism in male rats. Chem Biol Interact 177:142–152
Roth JA (2006) Homeostatic and toxic mechanisms regulating manganese uptake, retention, and elimination. Biol Res 39:45–57
Rouault TA, Tong WH (2008) Iron-sulfur cluster biogenesis and human disease. Trends Genet 24:398–407
Rousselet E, Moulis J-M (2008) Iron regulatory protein 1 is not an early target of cadmium toxicity in mice, but it is sensitive to cadmium stress in a human epithelial cell line. Biochem Cell Biol 86:416–424
Rousselet E, Martelli A, Chevallet M, Diemer H, Van Dorsselaer A, Rabilloud T, Moulis J-M (2008a) Zinc adaptation and resistance to cadmium toxicity in mammalian cells: molecular insight by proteomic analysis. Proteomics 8:2244–2255
Rousselet E, Richaud P, Douki T, Garcia Chantegrel J, Favier A, Bouron A, Moulis J-M (2008b) A zinc-resistant human epithelial cell line is impaired in cadmium and manganese import. Toxicol Appl Pharmacol 230:312–319
Ryu DY, Lee SJ, Park DW, Choi BS, Klaassen CD, Park JD (2004) Dietary iron regulates intestinal cadmium absorption through iron transporters in rats. Toxicol Lett 152:19–25
Sabolić I, Breljak D, Škarica M, Herak-Kramberger CM (2010) Role of metallothionein in cadmium traffic and toxicity in kidneys and other mammalian organs. Biometals. doi:10.1007/s10534-010-9351-z
Satarug S, Kikuchi M, Wisedpanichkij R, Li B, Takeda K, Na-Bangchang K, Moore MR, Hirayama K, Shibahara S (2008) Prevention of cadmium accumulation in retinal pigment epithelium with manganese and zinc. Exp Eye Res 87:587–593
Satarug S, Garrett SH, Sens MA, Sens DA (2010) Cadmium, environmental exposure, and health outcomes. Environ Health Perspect 118:182–190
Saydam N, Adams TK, Steiner F, Schaffner W, Freedman JH (2002) Regulation of metallothionein transcription by the metal-responsive transcription factor MTF-1: identification of signal transduction cascades that control metal-inducible transcription. J Biol Chem 277:20438–20445
Scheiber B, Goldenberg H (1993) NAD(P)H:ferric iron reductase in endosomal membranes from rat liver. Arch Biochem Biophys 305:225–230
Scott BJ, Bradwell AR (1983) Identification of the serum binding proteins for iron, zinc, cadmium, nickel, and calcium. Clin Chem 29:629–633
Sekler I, Sensi SL, Hershfinkel M, Silverman WF (2007) Mechanism and regulation of cellular zinc transport. Mol Med 13:337–343
Sensi SL, Paoletti P, Bush AI, Sekler I (2009) Zinc in the physiology and pathology of the CNS. Nat Rev Neurosci 10:780–791
Shoolingin-Jordan PM, Spencer P, Sarwar M, Erskine PE, Cheung KM, Cooper JB, Norton EB (2002) 5-Aminolaevulinic acid dehydratase: metals, mutants and mechanism. Biochem Soc Trans 30:584–590
Sikorski EM, Uo T, Morrison RS, Agarwal A (2006) Pescadillo interacts with the cadmium response element of the human heme oxygenase-1 promoter in renal epithelial cells. J Biol Chem 281:24423–24430
Singh KP, Kumari R, Pevey C, Jackson D, DuMond JW (2009) Long duration exposure to cadmium leads to increased cell survival, decreased DNA repair capacity, and genomic instability in mouse testicular Leydig cells. Cancer Lett 279:84–92
Smedman M, Potempska A, Rubenstein R, Ju W, Ramakrishna N, Denman RB (1997) Effects of cadmium, copper, and zinc and beta APP processing and turnover in COS-7 and PC12 cells. Relationship to Alzheimer disease pathology. Mol Chem Neuropathol 31:13–28
Sommer R, Beyersmann D (1984) Zinc and cadmium in 5-aminolevulinic acid dehydratase. Equilibrium, kinetic, and 113Cd-nmr-studies. J Inorg Biochem 20:131–145
Sopjani M, Foller M, Dreischer P, Lang F (2008) Stimulation of eryptosis by cadmium ions. Cell Physiol Biochem 22:245–252
Staessen JA, Buchet JP, Ginucchio G, Lauwerys RR, Lijnen P, Roels H, Fagard R (1996) Public health implications of environmental exposure to cadmium and lead: an overview of epidemiological studies in Belgium. J Cardiovasc Risk 3:26–41
Syapin PJ (2008) Regulation of haeme oxygenase-1 for treatment of neuroinflammation and brain disorders. Br J Pharmacol 155:623–640
Takeda K, Ishizawa S, Sato M, Yoshida T, Shibahara S (1994) Identification of a cis-acting element that is responsible for cadmium-mediated induction of the human heme oxygenase gene. J Biol Chem 269:22858–22867
Takenouchi T, Fujimoto M, Shimamoto A, Munekata E (1999) Isolation and characterization of Cox17p from porcine heart by determining its survival-promoting activity in NIH3T3 cells. Biochim Biophys Acta 1472:498–508
Thévenod F (2009) Cadmium and cellular signaling cascades: to be or not to be? Toxicol Appl Pharmacol 238:221–239
Thomas LD, Hodgson S, Nieuwenhuijsen M, Järup L (2009) Early kidney damage in a population exposed to cadmium and other heavy metals. Environ Health Perspect 117:181–184
Tselepis C, Ford SJ, McKie AT, Vogel W, Zoller H, Simpson RJ, Diaz-Castro J, Iqbal TH, Ward DG (2010) Characterisation of the transition metal binding properties of hepcidin. Biochem J 427:289–296
Tsiftsoglou AS, Vizirianakis IS, Strouboulis J (2009) Erythropoiesis: model systems, molecular regulators, and developmental programs. IUBMB Life 61:800–830
Vallee BL, Falchuk KH (1993) The biochemical basis of zinc physiology. Physiol Rev 73:79–118
van den Berghe PV, Klomp LW (2010) Posttranslational regulation of copper transporters. J Biol Inorg Chem 15:37–46
Van Kerkhove E, Swennen Q (2010) Cadmium and transport of ions and substances across cell membranes and epithelia. Biometals (this issue)
Vangheluwe P, Sepulveda MR, Missiaen L, Raeymaekers L, Wuytack F, Vanoevelen J (2009) Intracellular Ca2+- and Mn2+-transport ATPases. Chem Rev 109:4733–4759
Varela-Nallar L, Toledo EM, Larrondo LF, Cabral AL, Martins VR, Inestrosa NC (2006) Induction of cellular prion protein gene expression by copper in neurons. Am J Physiol Cell Physiol 290:C271–C281
Wallander ML, Leibold EA, Eisenstein RS (2006) Molecular control of vertebrate iron homeostasis by iron regulatory proteins. Biochim Biophys Acta 1763:668–689
Weiss JH, Sensi SL (2000) Ca2+-Zn2+ permeable AMPA or kainate receptors: possible key factors in selective neurodegeneration. Trends Neurosci 23:365–371
Wernimont AK, Huffman DL, Lamb AL, O’Halloran TV, Rosenzweig AC (2000) Structural basis for copper transfer by the metallochaperone for the Menkes/Wilson disease proteins. Nat Struct Biol 7:766–771
Wijesekara N, Chimienti F, Wheeler MB (2009) Zinc, a regulator of islet function and glucose homeostasis. Diabetes Obes Metab 11 Suppl 4: 202-14
Wills NK, Ramanujam VM, Kalariya N, Lewis JR, van Kuijk FJ (2008) Copper and zinc distribution in the human retina: relationship to cadmium accumulation, age, and gender. Exp Eye Res 87:80–88
Zhang B, Georgiev O, Hagmann M, Gunes C, Cramer M, Faller P, Vašák M, Schaffner W (2003) Activity of metal-responsive transcription factor 1 by toxic heavy metals and H2O2 in vitro is modulated by metallothionein. Mol Cell Biol 23:8471–8485
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moulis, JM. Cellular mechanisms of cadmium toxicity related to the homeostasis of essential metals. Biometals 23, 877–896 (2010). https://doi.org/10.1007/s10534-010-9336-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-010-9336-y