
Abstract
The recent rise in drug resistance found amongst community acquired infections has sparked renewed interest in developing antimicrobial agents that target resistant organisms and limit the natural selection of immune variants. Recent discoveries have shown that iron uptake systems in bacteria and fungi are suitable targets for developing such therapeutic agents. The use of siderophore-drug conjugates as “Trojan Horse” drug delivery agents has attracted particular interest in this area. This review will discuss efforts in our research group to study the salmycin class of “Trojan Horse” antibiotics. Inspired by the natural design of the salmycins, a series of desferridanoxamine-antibiotic conjugates were synthesized and tested in microbial growth inhibition assays. The results of these studies will be related to understanding the role of drug release in siderophore-mediated drug delivery with implications for future siderophore-drug conjugate design.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Antibiotic resistance is developing at an alarming rate. Cases of resistance have now been reported for all approved antibacterial drugs used in clinical settings. The gravity of this problem is becoming apparent as resistant infections once limited to hospitals are spreading to the general community (Marshall 2008). Some experts even foreshadow a fast return to the helpless pre-antibiotic days (Ash 1996; Mitscher 2008; Nathan 2004) as the pipeline of last resort drugs runs dry and antibiotic development amongst major pharmaceutical companies is at an all time low (Jarvis 2008).
The evolution of resistance to antibiotics is seemingly unavoidable. To regain the upper hand against resistant infections, modern antibiotic discovery programs should have at the fore front a goal of developing new antimicrobial agents that limit the emergence of resistance (Grand Challenges 2008). Our group and others have demonstrated that iron uptake systems in bacteria are suitable targets for developing such antibiotic therapies. For example, the use of bacterial siderophores as drug delivery agents can bypass membrane associated resistance mechanisms (such as decreased permeability barriers and efflux) and increase the potency of drugs up to 100-fold relative to passive diffusion (Braun 1999) while limiting the selection of resistance (by simultaneously targeting multiple pathogen components) to non-pathogenic mutants deficient in siderophore-receptor proteins. This review highlights siderophore-mediated drug delivery and addresses the effect of drug release on antibacterial activity of siderophore-antibiotic conjugates.
The siderophore iron uptake system
Iron is essential for nearly all forms of life and is a critical virulence factor during the course of a pathogenic infection. Although bacteria have evolved several methods for iron assimilation, the most common involves the production of siderophores, low molecular weight iron(III) chelators, for extracellular chelation followed by active transport into the cell (Guerinot 1994; Neilands 1995; Winkelmann and van der Helm 1987). The siderophore system becomes fully operational under times of iron restriction, such as during pathogen invasion of a host, and is initiated by the up-regulation of siderophore biosynthetic genes and the expression of siderophore receptor/transport proteins. Siderophores are excreted to the extracellular environment where they bind iron (III) and subsequently are recognized and transported into the cell by an intricate system of membrane-associated proteins (Krewulak and Vogel 2008). Most microbes produce ferric reductase enzymes that can then liberate the internalized iron and in most, but not all cases, the siderophore can reenter the catalytic cycle (Fig. 1). For more information on this highly studied process the reader is directed elsewhere (Ratledge and Dover 2000).

General model of siderophore-mediated iron acquisition in bacteria
Siderophore-mediated iron acquisition is essential for the survival of pathogenic bacteria. For this reason, siderophore iron uptake systems have been identified as suitable targets for antimicrobial therapies (Miethke and Marahiel 2007). Three approaches currently being pursued in this area include: (1) iron starvation via competitive chelation (Miller et al. 2009), (2) inhibition of siderophore biosynthesis (Ferreras et al. 2005; Neres et al. 2008; Quadri 2007), and (3) siderophore-mediated drug delivery (“Trojan Horse” antibiotics). Our research group is involved in all three areas of exploiting iron assimilation for antibiotic development, but this review will focus on our recent efforts to study siderophore-mediated drug delivery.
Siderophore-mediated drug delivery (“Trojan Horse” antibiotics)
Bacteria express siderophore receptor proteins that specifically recognize the iron complexes of their native sideorphores. To ensure a competitive growth advantage bacteria commonly possess the machinery to recognize and transport siderophore-iron complexes produced by other species (Challis and Hopwood 2003). Along this evolutionary pathway certain bacteria have also learned to exploit this “iron thievery” for antibiotic delivery by attaching toxic substances to siderophores. Some of these naturally occurring siderophore-drug conjugates, also referred to as sideromycins, include the danomycins (Huber et al. 1986), salmycins (Fig. 2) (Vértesy et al. 1995), albomycins (Fig. 2) (Benz et al. 1982; Gause 1955), ferrimycins (Bickel et al. 1960, 1965; Sackmann et al. 1962), and microcins (Thomas et al. 2004).

Representative examples of siderophore-drug conjugates
Inspired by nature’s design our group and others have demonstrated that rationally designed synthetic siderophore-drug conjugates can also be used as effective drug delivery systems. The use of siderophore-drug conjugates as “Trojan Horse” antibiotics has been reviewed extensively (Miller et al. 2009; Miller and Malouin 1993; Miethke and Marahiel 2007; Möllmann et al. 2009; Roosenberg et al. 2000). This topic has maintained high interest amid current research programs (Braun et al. 2008; Miller et al. 2009; Möllmann et al. 2009; Nolan and Walsh 2008; Rivault et al. 2007; Wach et al. 2008; Rook 2008).
Although much is already known about siderophore-drug conjugates, a more detailed understanding of how this delivery system works will facilitate the ultimate goal of designing more effective conjugates. As summarized in Fig. 3, iron-complexed siderophore-drug conjugates are transported into bacterial cells via siderophore receptor/transport proteins. To be active, the drug still has to be able to reach its known target. However, whether the conjugate itself is the active compound, or if drug release is necessary for activity is not fully understood.

General model of siderophore-mediated drug delivery in bacteria
Is drug release necessary for antimicrobial activity of siderophore-drug conjugates?
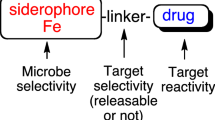
Siderophore-drug conjugates are three component systems consisting of a siderophore, linker, and drug (Figs. 2, 3). All three components serve an important function. The role of the siderophore is well understood (see earlier “Discussion”), but less attention has been given to the nature of the drug and the stability of the linker. The drug (structure activity relationships and target location) determines if drug release is necessary for activity while the linker determines if this release is possible in cells.
Many examples of β-lactam antibiotics with iron binding side chains and rationally designed siderophore-β-lactam conjugates with potent antimicrobial activity have been reported (Miller et al. 2009; Heinisch et al. 2003, 2002; Wittmann et al. 2002; Roosenberg et al. 2000). Beta-lactam antibiotics tolerate significant side chain modification with regards to the affinity for their target penicillin binding proteins (PBPs), which are located in the periplasm. Competitive binding studies with PBPs proved that the entire conjugates are capable of binding to the target. Outer membrane protein analyses unambiguously proved that entry into bacterial cells is dependent on siderophore-iron uptake systems and resistance always resulted in deletion of the outer membrane siderophore-receptor proteins. No cross-resistance to the parent β-lactam drug (modification of PBPs) was observed. These studies suggest that these conjugates only require transport to the periplasm to reach the target PBPs and β-lactam drugs might not require release for activity (Brochu et al. 1992).
Although there are only a limited number of examples available in the literature (Hennard et al. 2001; Rivault et al. 2007; Zähner et al. 1977), nearly all siderophore-drug conjugates featuring antibiotic components that have cytoplasmic targets show decreased activity compared to the free drug alone. For drugs with cytoplasmic targets it appears that linkage to siderophores either inhibits the drug from reaching its target or causes a significant decrease in binding affinity to the target, both of which lead to loss of activity. The only example of such a conjugate which retained or exceeded the activity of the parent drug involved the use of pyoverdine, a siderophore produced by P. aeruginosa, coupled to quinolone antibiotics using a labile linker (Fig. 2). The use of a labile linker was critical for increasing activity of the conjugate relative to the free drug alone (Hennard et al. 2001; Rivault et al. 2007). However, P. aeruginosa is a special case in that it has an exceptionally high outer membrane exclusion barrier (Nikaido et al. 1991) which is responsible for its high resistance to small molecule antibiotics. This makes P. aeruginosa an ideal candidate for siderophore-mediated drug delivery as reflected by the many successful applications of this targeted therapy (Budzikiewicz 2001; Heinisch et al. 2003, 2002; Hennard et al. 2001; Rivault et al. 2007; Wittmann et al. 2002). Although the pyoverdine–quinolone conjugates featuring a labile linker show promise in delivering and releasing quinolone antibiotics, it is still left to question if the drug is selectively released only inside the bacterial cell and how applicable this approach will be to other species of bacteria.
Nature seems to have already addressed these questions/problems by designing the albomycins (Fig. 2). The albomycins enter bacterial cells via the ferric hydroxamate transport systems (Hartmann et al. 1979; Pramanik and Braun 2006). The ferrichrome-like siderophore portion of the albomycins is recognized as the iron complex and transported by the ferrichrome associated proteins FhuA (Ferguson et al. 2000) and FhuD (Clarke et al. 2002) and once internalized the toxic thionucleoside is enzymatically released by a serine protease (Braun et al. 1983). The albomycins are extremely potent siderophore-drug conjugates active against Gram-positive and Gram-negative bacteria in vitro and in vivo (Braun et al. 2008; Gause 1955; Pramanik et al. 2007).
A close relative of the albomycins is the salmycin class of naturally occurring “Trojan Horse” antibiotics (Fig. 2) isolated from Streptomyces violaceus DSM 8286 that exhibit potent antibacterial activity (MIC of 10 nM) against Staphylococci and Streptococci including resistant strains (Vértesy et al. 1995). Similar to the albomycins, the salmycins appear to enter cells via ferric hydroxamate transport systems (Pramanik and Braun 2006). Originally it was proposed that the amino glycoside antibiotic is released upon hydrolysis inside cells (Vértesy et al. 1995). We hypothesize that iron reduction may trigger drug release via an intramolecular cyclization process as shown in Scheme 1 (Roosenberg and Miller 2000).

Proposed drug release mechanism of the salmycins (Roosenberg and Miller 2000)
With the ultimate goal of testing this hypothesis and evaluating the scope of desferridanoxamine as a vector for drug delivery we synthesized desferridanoxamine (Dan), desferrisalmycin B (Sal B), and a variety of desferridanoxamine-antibiotic conjugates utilizing antibiotics with drastically different cellular targets. Biological activity was evaluated using a variety of bacterial strains summarized in Table 1.
Synthesis and biological activity of desferridanoxamine
Danoxamine (Fig. 2) was first isolated and characterized as the siderophore component of the sideromycin antibiotic danomycin (Huber et al. 1986) and later rediscovered as the iron binding component of the salmycins (Vértesy et al. 1995). Desferridanoxamine (Dan) is a linear tridentate siderophore comprised of three sequential hydroxamic acid (Codd 2008) iron binding groups. When viewed in a retrosynthetic manner (Scheme 2), disconnecting the siderophore backbone at the hydroxamic acid and amide bonds revealed two major components, derived from synthetic intermediates 7 and 3, joined by repeating succinyl linkers (Roosenberg and Miller 2000). The synthesis relied on the production of protected precursor 12 which allowed for handling and derivatization of the siderophore without fear of iron contamination or competitive reactivity of the nucleophilic hydroxamates. This was important for providing synthetic access to the salmycins and other desferridanoxamine conjugates (as will be highlighted later).

Retrosynthetic analysis of desferridanoxamine (Dan) (Roosenberg and Miller 2000)
Tetra-O-benzyldanoxamine (12) was prepared on multi-gram scale (Long T. E., manuscript in preparation) following an 11-step convergent synthesis starting from O-benzylhydroxylamine HCl (1) and 1, 5-pentanediol (5) (Scheme 3) (Roosenberg and Miller 2000). Some key features of the synthesis include use of benzyloxy-carbamate 2 as a “hydroxamate equivalent” that is incorporated into alkyl units via deprotonation and SN2 displacement of a leaving group, use of nitrile 3 as a protected primary amine that is revealed using Raney nickel reduction following the Bergeron protocol (Bergeron and Pegram 1988), and sequential N-Boc deprotection, succinic anhydride acylation, and diphenylphosphoryl azide (DPPA) amide coupling used to assemble the fully protected scaffold 12. This synthetic route allowed for rapid synthesis of 12 in relatively high yield (up to 9% yield for the longest linear sequence) on large scale (~5 g). Desferridanoxamine was obtained in excellent purity and high yield upon global deprotection of 12 via Pd-catalyzed hydrogenation (Scheme 3).

Total synthesis of desferridanoxamine (Roosenberg and Miller 2000). Multi-gram scale synthesis of tetra-O-benzyldanoxamine precursor 12 (Long T. E., manuscript in preparation)
Growth promotion studies using desferridanoxamine (Dan) relative to ferricrocin (F) and mycobactin J (M) were performed on iron-restricted media (Schumann and Möllmann 2001) to determine which species can utilize desferridanoxamine for iron assimilation (Table 2). Desferridanoxamine effectively promoted the growth of Gram-positive (S. aureus and B. subtilis), select Gram-negative (P. aeruginosa and S. typhimurium), and select mycobacterium (M. smegmatis) strains. Interestingly, no E. coli strains were able to utilize desferridanoxamine. All Gram-negative and Gram-positive strains were able to utilize ferricrocin except E. coli strains BR158 and HK9/7 which lack essential TonB and FhuA/FhuE, respectively. Only mycobacteria strains M. smegmatis mc2155 and Mc2155-M24 were able to use desferridanoxamine for growth promotion. All mycobacteria mutants deficient in the biosynthesis and/or uptake of exochelin were not able to utilize desferridanoxamine. Possibly danoxamine can undergo iron exchange with exochelin and act as a growth promoter independent of the presence of mycobactin. All mycobacteria strains were able to utilize mycobactin J for growth promotion. Full details of growth promotion studies will be reported soon (Long T. E., manuscript in preparation).
Synthesis and biological activity of desferrisalmycin B
We recently reported the first total synthesis and stereochemical assignment of desferrisalmycin B (Sal B) and epi-desferrisalmycin B (epi -Sal B) (Dong et al. 2002). The unique structure of the salmycins (Fig. 2) includes an iron-binding siderophore (danoxamine) and an unusual amino-disaccharide antibiotic component. From a retrosynthetic standpoint we envisioned making two major disconnections at the ester and glycosidic linkages (Scheme 4). This approach required the syntheses of three major components to be assembled in a convergent fashion (Scheme 5): (1) tetra-O-benzyldanoxamine (12), (2) glycosyl acceptor fragment 13, and (3) glycosyl donor fragment 14 (Dong et al. 2002).

Retrosynthetic analysis of desferrisalmycin B (Sal B) (Dong et al. 2002)

Total synthesis of desferrisalmycin B (Sal B) and epi-desferrisalmycin B (epi -Sal B) (Dong et al. 2002)
Tetra-O-benzyldanoxamine (12) was synthesized as described earlier in Scheme 2 (Roosenberg and Miller 2000). The syntheses of the glycosyl acceptor (13) and donor (14) fragments as well as the final assembly of all the key fragments are shown in Scheme 5. Trichloroacetimidate 13 was chosen as the glycosyl acceptor fragment and was synthesized efficiently from d-mannose on large scale. The synthesis of the novel glycosyl donor fragment 14 featured a key stereoselective dihydroxylation of olefin 18 to give mono-protected diol 19. Both epimers (19 and epi -19) of C-6” were available (via Mitsunobu-mediated stereochemical inversion) in high enantiomeric purity (absolute stereochemistry confirmed using correlation studies). A Bose-modified Mitsunobu reaction gave azide 20 (epi -20) which allowed straight forward access to glycosyl donor 21 (epi -21). The O-trimethylsilyl triflate-mediated stereoselective glycosylation reaction between 13 and 21 (epi -21) proceeded smoothly in high yield (88%) to give protected disaccharide 22 (epi -22) with high enantiomeric purity. Unfortunately, after deprotection of the TBS protected primary alcohol of 22 (epi -22) direct couplings with 12 failed under a variety of conditions. To circumvent this problem the succinyl linker was installed on the disaccharide using a five-step protecting group manipulation sequence leading to 23 (epi -23). This carboxylic acid proved suitable for EDC/HOAt-mediated coupling to the O-benzyl hydroxylamine generated from 11. Fully protected desferrisalmycin B precursor 24 (epi -24) was then universally deprotected using an optimized hydrogenation reaction catalyzed by palladium black and perchloric acid to separately give both epimers of desferrisalmycin B (25 and epi -25). Comparison of 1H- and 13C-NMR data to authentic material revealed that epi -25 is the natural desferrisalmyin B and the heptopyranose component has a d-glycero-d-gluco configuration (Dong et al. 2002).
As expected there was no difference in the antibacterial activities of the synthetic or naturally derived salmycins (Tables 3, 4). Some general observations include that the activity of salmycin A (Sal A) was superior to the activity of albomycin (Alb) and desferrisalmycin B. There was a noticeable reduction in activity by conversion of desferrisalmycin B to epi-desferrisalmycin B supporting the potential existence of a distinct cellular target for the novel amino-disaccharide antibiotic, which is believed to be an inhibitor of protein synthesis (Braun et al. 2008). Compared to albomycin, the salmycins had greater activity against multidrug resistant S. aureus (MRSA), especially with efflux-mediated multiresistance (ERSA), where albomycin showed no activity. Albomycin was effective against Gram-positive and Gram-negative bacteria while the salmycins were primarily effective only against Gram-positive organisms (except for the activity of salmycin A against K. pneumoniae). Despite the strong species-selective antimicrobial activity of the salmycins and albomycin, rapid development of resistant mutants under in vitro conditions was observed as seen in Fig. 4 (Möllmann et al. 2004).

Inhibition zones of ciprofloxacin (Cip), epi-desferrisalmycin (epi -Sal B), desferrsalmycin B (Sal B), desferridanoxamine (Dan), salmycin A (Sal A), albomycin (Alb), and kanamycin (Kan) against a S. aureus SG 511 b S. aureus 134/93 c S. aureus 994/93 d S. aureus EfS 4. Exactly 50 μl of a 100 mg/l solution of each compound was filled in 9 mm wells in Mueller-Hinton agar. Pictures taken after incubation at 37°C for 24 h. Resistant colonies can be seen as white dots within the inhibition zones
The convergent approach taken in Scheme 5 allows for synthesis of all the members of the salmycin class and production of unique analogs. Having complete control over the covalent structure presents the opportunity for studying structure activity relationships of the salmycins and independently the amino-disaccharide component, a potentially novel small molecule antibiotic. Additionally, novel analogs can be synthesized that impart desirable properties (improved pharmacokinetics), probe mechanism of action, and test our hypothesized reductase-triggered, intramolecular drug release process (Scheme 1).
Synthesis and biological activity of desferridanoxamine-antibiotic conjugates
To probe the drug delivery capabilities of the salmycins we synthesized a series of desferridanoxamine-antibiotic conjugates and evaluated their activity in growth inhibition assays. We chose a variety of commercially available antibiotics with drastically different cellular targets (Fig. 5). The conjugates were prepared using direct active ester couplings on the fully protected precursor 12 followed by global deprotection. The optimized conditions are shown in Scheme 6 while full experimental details will be published elsewhere (Long T. E., manuscript in preparation). The couplings all proceeded in high yield and gave desirable amounts of the fully protected conjugates (typically~100 mg). Benzyl protection of the hydroxamates facilitated chromatographic purification without the fear of iron contamination. The final hydrogenation of this pure material provided the iron-free conjugates (fully characterizable by NMR) in good yield and high purity.

Antibiotics selected for desferridanoxamine conjugates (Walsh 2003)

Synthesis of desferridanoxamine-antibiotic conjugates (Long T. E., manuscript in preparation)
The desferridanoxamine-antibiotic conjugates were evaluated for their ability to inhibit the growth of bacteria using the agar well diffusion test (Table 5) relative to the free siderophore, desferridanoxamine (Dan), and the free drug, Lorabid® (Lor), ciprofloxacin (Cip), or triclosan (Tri). As expected, all of the conjugates demonstrated growth inhibitory activity. Unexpectedly, desferridanoxamine, the free siderophore, demonstrated growth inhibitory activity against all E. coli strains and M. vaccae. This was consistent with lack of growth promotion (Table 2) and could be a case of iron limitation considering the relatively high concentration (2.0 mM) of desferridanoxamine used in the assay. The rational for these strains is also supported by the fact that the desferridanoxamine-Lorabid® conjugate (Dan-Lor) and the desferridanoxamine-ciprofloxacin conjugate (Dan-Cip) seemed to mimic the activity displayed by desferridanoxamine alone. The desferridanoxamine-Lorabid® conjugate reflected activity observed for previously studied siderophore-Lorabid® conjugates (Roosenberg et al. 2000) in that it hit the same panel of organisms as the parent drug, but showed decreased activity. The desferridanoxamine-ciprofloxacin conjugate showed decreased activity in all cases compared to ciprofloxacin alone. Since we know that desferridanoxamine is recognized and actively transported by a number of the strains, the diminished activity of the conjugate implies that the quinolone drug either cannot reach its target or has a decreased affinity for its target. The activity of these conjugates (Dan-Lor and Dan-Cip) is reduced compared to the free drug because there is probably no release of the drug.
The most active conjugate was the desferridanoxamine-triclosan conjugate (Dan-Tri). This conjugate exhibits equal or greater activity than the free drug alone for almost all the organisms screened. The MIC data (Table 6) for Dan-Cip and Dan-Tri relative to Cip and Tri appear to parallel the findings observed during the agar diffusion assay. The amide linked conjugates were less active than the free drug alone while the phenolic ester linked conjugate showed equal or great activity. The increased hydrolytic lability of the phenolic ester of Dan-Tri relative to the amide bonds of Dan-Lor and Dan-Cip implies that the free triclosan drug might have been released prematurely in the assay media, as supported by retained activity against E. coli strains unable to utilize desferridanoxamine for growth promotion (Tables 2, 5, 6). Still left for questioning is the possibility of intramolecular hydroxamate assisted release of triclosan (Scheme 1) especially since the fully benzyl protected triclosan conjugate (28) proved to be stable in the media (virtually no growth inhibitory activity was observed for 28, data not shown).
Further studies of the relationship between growth inhibitory activity of the conjugates and the stability of the drug linkage are merited. Efforts are underway to investigate the mode of action of these conjugates (determine if conjugate influx takes place via ferric hydroxamate transport) and evaluate the relationship between ferric ion concentration and the activity of the conjugates. Preliminary studies using salmycin resistant mutants of S. aureus and M. luteus suggest that resistance to salmycins correlates directly with resistance to desferridanoxamine-antibiotics (data not shown). Since the antibiotics have drastically different targets, cross-resistance supports entrance of the conjugates via siderophore influx mechanisms and not passive diffusion. Full reports of these studies will be published in the near future.
Summary and conclusions
The total syntheses of desferridanoxamine and desferrisalmycin B have recently been accomplished in our laboratories (Roosenberg and Miller 2000; Dong et al. 2002). Desferridanoxamine is an effective growth promoter for Gram-negative and Gram-positive bacteria. Desferrisalmycin B exhibits potent growth inhibitory activity against Gram-positive organisms including resistant strains. Using synthetic protocols developed during this work, a new series of desferridanoxamine conjugates of commercial antibiotics were prepared to investigate the drug transporting ability of desferridanoxamine and the drug release hypothesis of the salmycins (Long T. E., manuscript in preparation). These conjugates appear to be recognized and actively transported into bacterial cells. Evaluation of antibacterial activity of all the conjugates revealed that amide linked conjugates are less active than the free drug alone while the ester linked conjugate has equal or greater activity compared to the free drug alone. This data suggests that to optimize potency an active drug release process is needed for siderophore-drug conjugates utilizing drugs with intracellular targets.
The mechanism of drug release of the salmycins is still unknown, but will be the focus of future research. With antibiotic resistance on the rise and antibiotic development on the decline there is a serious need for new antimicrobial agents that limit the development of resistance. With detailed attention to proper design, siderophore-drug conjugates show promise as drug delivery agents that limit the formation of resistance and bypass known resistance mechanisms (decreased membrane permeability and efflux). Particular attention should be given to the choice of drugs (target location) and linkers (drug releasability). A better understanding of the role drug release plays in the activity of siderophore-drug conjugates will aid in the future design of more effective “Trojan Horse” antibiotics.
References
Ash C (1996) Antibiotic resistance: the new apocalypse? Trends Microbiol 4:371–372. doi:10.1016/0966-842X(96)30028-0
Benz G, Schröder T, Kurz J, Wünsche C, Karl W, Steffen GJ, Pfitzner J, Schmidt D (1982) Konstitution der deferriform der albomycine δ1, δ2 und ε. Angew Chem Int Ed 94:552–553
Bergeron RJ, Pegram JJ (1988) An efficient total synthesis of desferrioxamine B. J Org Chem 53:3131–3134. doi:10.1021/jo00249a001
Bickel H, Gäumann E, Nussberg G, Reusser P, Vischer E, Voser W, Wettstein A, Zähner H (1960) Stoffwechselprodukte von Actinomyceten. 25 Mitteilung: über die isolierung und charakterisierung der ferrimycine A1 und A2, neuer antibiotika der sideromycin-gruppe. Helv Chim Acta 43:2105–2118. doi:10.1002/hlca.19600430730
Bickel H, Mertens P, Prelog V, Seibl J, Walser A (1965) Constitution of ferrimycin A1. Antimicrob Agents Chemother 5:951–957
Braun V (1999) Active transport of siderophore-mimicking antibacterials across the outer membrane. Drug Resist Updat 2:363–369. doi:10.1054/drup.1999.0107
Braun V, Günthner H, Hantke K, Zimmerman L (1983) Intracellular activation of albomycin in Escherichia coli and Salmonella typhimurium. J Bacteriol 156:308–315
Braun V, Pramanik A, Gwinner T (2008) Use of sideromycins as tools and antibiotics. Paper presented at the 6th international biometals symposium, Santiago de Compostela, Spain, 14–18 July 2008
Brochu A, Brochu N, Nicas TI, Parr TR, Minnick AA, Dolence EK, McKee JA, Miller MJ, Lavoie MC, Malouin F (1992) Modes of action and inhibitory activities of new siderophore-β-lactam conjugates that use specific iron uptake pathways for entry into bacteria. Antimicrob Agents Chemother 36:2166–2175
Budzikiewicz H (2001) Siderophore-antibiotic conjugates used as trojan horses against Pseudomonas aeruginosa. Curr Top Med Chem 1:73–82. doi:10.2174/1568026013395524
Challis GL, Hopwood DA (2003) Synergy and contingency as driving forces for the evolution of multiple secondary metabolite production by Streptomyces species. Proc Natl Acad Sci USA 100:14555–14561. doi:10.1073/pnas.1934677100
Clarke TE, Braun V, Winkelmann G, Tari LW, Vogel HJ (2002) X-ray crystallographic structures of the Eschericia coli periplasmic protein FhuD bound to hydroxamate-type siderophores and the antibiotic albomycin. J Biol Chem 277:13966–13972. doi:10.1074/jbc.M109385200
Codd R (2008) Traversing the coordination chemistry and chemical biology of hydroxamic acids. Coord Chem Rev 252:1387–1408. doi:10.1016/j.ccr.2007.08.001
Dong L, Roosenberg JM, Miller MJ (2002) Total synthesis of deferrisalmycin B. J Am Chem Soc 124:15001–15005. doi:10.1021/ja028386w
Ferguson AD, Braun V, Fiedler HP, Coulton JW, Diederichs K, Welte W (2000) Crystal structure of the antibiotic albomycin in complex with the outer membrane transporter FhuA. Protein Sci 9:956–963
Ferreras JA, Ryu JS, Di Lello F, Tan DS, Quadri LEN (2005) Small molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat Chem Biol 1:219–232. doi:10.1038/nchembio706
Gause GF (1955) Recent studies on albomycin, a new antibiotic. BMJ 12:1177–1179
Guerinot ML (1994) Microbial iron transport. Annu Rev Microbiol 48:743–772. doi:10.1146/annurev.mi.48.100194.003523
Hartmann A, Fiedler HP, Braun V (1979) Uptake and conversion of the antibiotic albomycin by Eschericia coli K-12. Eur J Biochem 99:517–524. doi:10.1111/j.1432-1033.1979.tb13283.x
Grand Challenges in Global Health (2008) Create drugs and delivery systems to limit drug resistance. http://www.gcgh.org/LimitDrugResistance/Topics/DoNotGenerateResistance/Pages/default.aspx. Cited 11 Nov 2008
Heinisch L, Wittmann S, Stoiber T, Berg A, Ankel-Fuchs D, Möllmann U (2002) Highly antibacterial active amnioacyl penicillin conjugates with bis-catecholate siderophores based on secondary diamino acids and related compounds. J Med Chem 45:3032–3040. doi:10.1021/jm010546b
Heinisch L, Wittmann S, Stoiber T, Scherlitz-Hoffmann I, Ankel-Fuchs D, Möllmann U (2003) New tris- and tetrakis-catecholate siderophores based on polyazaalkanoic acids and their β-lactam conjugates. Arzneim-Forschung Drug Res 53:188–195
Hennard C, Truong QC, Desnottes JF, Paris JM, Moreau NJ, Abdallah MA (2001) Synthesis and activities of pyoverdin–quinolone adducts: a prospective approach to a specific therapy against Pseudomonas aeruginosa. J Med Chem 44:2139–2151. doi:10.1021/jm990508g
Ho WH, Wong HNC (1995) Chiral liquid crystalline compounds from d-(+)-glucose. Tetrahedron 51:7373–7388. doi:10.1016/0040-4020(95)00386-M
Huber P, Leuenberger H, Keller-Schierlein W (1986) Danoxamin, der eisenbindende teil des sideromycin-antibioticums danomycin. Helv Chim Acta 69:236–245. doi:10.1002/hlca.19860690128
Jarvis LM (2008) An uphill battle. C E News 86:15–20
Klare I, Heier H, Claus H, Reissbrodt R, Witte W (1995) VanA-mediated high-level glycopeptides resistance in Enterococcus faecium from animal husbandry. FEMS Microbiol Lett 125:165–172. doi:10.1111/j.1574-6968.1995.tb07353.x
Krewulak KD, Vogel HJ (2008) Structural biology of bacterial iron uptake. Biochim Biophys Acta 1778:1781–1804. doi:10.1016/j.bbamem.2007.07.026
Marshall E (2008) The bacteria fight back. Science 321:356–364. doi:10.1126/science.321.5887.356
Miethke M, Marahiel MA (2007) Siderophore-based iron acquisition and pathogen control. Microbiol Mol Biol Rev 71:413–451. doi:10.1128/MMBR.00012-07
Miller MJ, Malouin F (1993) Microbial iron chelators as drug delivery agents: the rational design and synthesis of siderophore-drug conjugates. Acc Chem Res 26:241–249. doi:10.1021/ar00029a003
Miller MJ, Zhu H, Xu Y, Wu C, Walz AJ, Vergne A, Roosenberg JM, Moraski G, Minnick AA, McKee-Dolence J, Hu J, Fennell K, Dolence EK, Dong L, Franzblau S, Malouin F, Möllmann U (2009) Utilization of microbial iron assimilation processes for the development of new antibiotics and inspiration for the design of new anticancer agents. Biometals 22:61–75
Mitscher LA (2008) Coevolution: mankind and microbes. J Nat Prod 71:497–509. doi:10.1021/np078017j
Möllmann U, Dong L, Vertesy L, Miller MJ (2004) Salmycins—natural siderophore-drug conjugates: Prospects for modification and investigation based on successful total synthesis. Paper presented at the 2nd international Biometals symposium, Garmisch-Partenkirchen, Germany, 3–5 Sept 2004
Möllmann U, Heinisch L, Bauernfeind A, Thilo K, Ankel-Fuchs D (2009) Siderophores as drug delivery agents: application of the “Trojan Horse” strategy. Biometals. doi:10.1007/s10534-009-9219-2
Nathan C (2004) Antibiotics at the crossroads. Nature 431:899–902. doi:10.1038/431899a
Neilands JB (1995) Siderophores: structure and function of microbial iron transport compounds. J Biol Chem 270:26723–26726
Neres J, Labello NP, Somu RV, Boshoff HI, Wilson DJ, Vannada J, Chen L, Barry CE, Bennet EM, Aldich CC (2008) Inhibition of siderophore biosynthesis in Mycobacterium tuberculosis with nucleoside bisubstrate analogues: structure–activity relationships of the nucleobase domain of 5′-O-[N-(salicyl)sulfamoyl]adenosine. J Med Chem 51:5349–5370. doi:10.1021/jm800567v
Nikaido H, Nikaido K, Harayama S (1991) Identification and characterization of porins in Pseudomonas aeruginosa. J Biol Chem 266:770–779
Nolan EM, Walsh CT (2008) Investigations of the MceIJ-catalyzed posttranslational modification of the microcin E492 C-terminus: linkage of ribosomal and nonribosomal peptides to form “Trojan Horse” antibiotics. Biochemistry 47:9289–9299. doi:10.1021/bi800826j
Ogawa T, Kaburagi T (1982) Synthesis of a branched d-glucotetraose, the repeating unit of the extracellular polysaccharides of Grifola umbellate, Sclerotinia libertiana, Porodisculus pendulus, and Schizophyllum commune fries. Carbohydr Res 103:53–64. doi:10.1016/S0008-6215(82)80007-4
Pramanik A, Braun V (2006) Albomycin uptake via a ferric hydroxamate transport system of Streptococcus pneumoniae R6. J Bacteriol 188:3878–3886. doi:10.1128/JB.00205-06
Pramanik A, Stroeher UH, Krejci J, Standish AJ, Bohn E, Paton JC, Autenrieth IB, Braun V (2007) Albomycin is an effective antibiotic, as exemplified with Yersinia enterocolitica and Streptococcus pneumoniae. Int J Med Microbiol 297:459–469. doi:10.1016/j.ijmm.2007.03.002
Quadri LEN (2007) Strategic paradigm shifts in the antimicrobial drug discovery process of the 21st century. Infect Disord Drug Targets 7:230–237. doi:10.2174/187152607782110040
Ratledge CL, Dover G (2000) Iron metabolism in pathogenic bacteria. Annu Rev Microbiol 54:881–941. doi:10.1146/annurev.micro.54.1.881
Richmond MH, Clark DC, Wotton S (1976) Indirect method for assessing the penetration of beta-lactamase-nonsusceptible penicillins and cephalosporins in Eschericia coli. Antimicrob Agents Chemother 10:215–218
Rivault F, Liébert C, Burger A, Hoegy F, Abdallah MA, Schalk IJ, Mislin GLA (2007) Synthesis of pyochelin–norfloxacin conjugates. Bioorg Med Chem Lett 17:640–644. doi:10.1016/j.bmcl.2006.11.005
Rook G (2008) Anti-mycobacterial mycobactin-linked glyconanoparticles. In: Grants awarded. Grand Challenges in Global Health. http://www.gcgh.org/explorations/Pages/GrantsAwarded.aspx. Cited 11 Nov 2008
Roosenberg JM, Miller MJ (2000) Total synthesis of the siderophore danoxamine. J Org Chem 65:4833–4838. doi:10.1021/jo000050m
Roosenberg JM, Lin Y-M, Lu Y, Miller MJ (2000) Studies and syntheses of siderophores, microbial iron chelators, and analogs as potential drug delivery agents. Curr Med Chem 7:159–197
Sackmann W, Preusser P, Neipp L, Kradolfer F, Gross F (1962) Ferrimycin A, a new iron containing antibiotic. Antibiot Chemother 12:34–45
Schumann G, Möllmann U (2001) A screening system for xenosiderophores as potential drug delivery agents in mycobacteria. Antimicrob Agents Chemother 45:1317–1322. doi:10.1128/AAC.45.5.1317-1322.2001
Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR (1990) Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol Microbiol 4:1911–1919. doi:10.1111/j.1365-2958.1990.tb02040.x
Thomas X, Destoumieux-Garzón D, Peduzzi J, Afonso C, Blond A, Birlirakis N, Goulard C, Dubost L, Thai R, Tabet JC, Rebuffat S (2004) Siderophore peptide, a new type of post-translationally modified antibacterial peptide with potent activity. J Biol Chem 279:28233–28242. doi:10.1074/jbc.M400228200
Vértesy L, Aretz W, Fehlhaber H-W, Koger H (1995) Salmycin A–D, Antibiotika aus Streptomycese violaceus, DSM 8286, mit siderophore-aminoglycosid-struktur. Helv Chim Acta 78:46–60. doi:10.1002/hlca.19950780105
Wach JY, Bonazzi S, Gademann K (2008) Antimicrobial surfaces through natural product hybrids. Angew Chem Int Ed 47:7123–7126. doi:10.1002/anie.200801570
Walsh C (2003) Antibiotics: actions, origins, resistance. ASM Press, Washington, DC
Winkelmann G, van der Helm D, Neilands JB (1987) Iron transport in microbes, plants, and animals. VCH Press, Weinheim, pp 1–533
Witte W, Cuny C, Braulke C, Heuck D (1994) Clonal dissemination of two MRSA strains in Germany. Epidemiol Infect 113:67–73
Wittmann S, Schnabelrauch M, Scherlitz-Hoffmann I, Möllmann U, Ankel-Fuchs D, Heinisch L (2002) New synthetic siderophores and their β-lactam conjugates based on amino acids and dipeptides. Bioorg Med Chem 10:1659–1670. doi:10.1016/S0968-0896(02)00044-5
Zähner H, Diddens H, Keller-Schierlein W, Nägeli HU (1977) Some experiments with semisynthetic sideromycins. Jpn J Antibiot 30:S201–S206
Zimmermann W (1980) Penetration of β-lactam antibiotics into their target enzymes in Pseudomonas aeruginosa: comparison of a highly sensitive mutant with its parent strain. Antimicrob Agents Chemother 18:94–100
Acknowledgments
We gratefully acknowledge the National Institutes of Health (NIH) research grants RO1 AI054193, NIH AI 030988, and NIH GM025845 for financial support. We thank Irmgard Heinemann and Uta Wohlfeld for their excellent technical assistance with growth promotion and growth inhibition assays at the HKI. MJM gratefully acknowledges the kind hospitality of the HKI and the University of Notre Dame for a sabbatical opportunity in Jena, Germany. TAW gratefully acknowledges the University of Notre Dame Chemistry-Biochemistry-Biology (CBBI) Interface Program and NIH training grant T32GM075762 for a fellowship and the kind hospitality of the HKI for a research internship opportunity.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wencewicz, T.A., Möllmann, U., Long, T.E. et al. Is drug release necessary for antimicrobial activity of siderophore-drug conjugates? Syntheses and biological studies of the naturally occurring salmycin “Trojan Horse” antibiotics and synthetic desferridanoxamine-antibiotic conjugates. Biometals 22, 633–648 (2009). https://doi.org/10.1007/s10534-009-9218-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-009-9218-3