
Abstract
P1B-type ATPases transport heavy metals (Cu+, Cu2+, Zn2+, Co2+, Cd2+, Pb2+) across membranes. Present in most organisms, they are key elements for metal homeostasis. P1B-type ATPases contain 6-8 transmembrane fragments carrying signature sequences in segments flanking the large ATP binding cytoplasmic loop. These sequences made possible the differentiation of at least four P1B-ATPase subgroups with distinct metal selectivity: P1B-1: Cu+, P1B-2: Zn2+, P1B-3: Cu2+, P1B-4: Co2+. Mutagenesis of the invariant transmembrane Cys in H6, Asn and Tyr in H7 and Met and Ser in H8 of the Archaeoglobus fulgidus Cu+-ATPase has revealed that their side chains likely coordinate the metals during transport and constitute a central unique component of these enzymes. The structure of various cytoplasmic domains has been solved. The overall structure of those involved in enzyme phosphorylation (P-domain), nucleotide binding (N-domain) and energy transduction (A-domain), appears similar to those described for the SERCA Ca2+-ATPase. However, they show different features likely associated with singular functions of these proteins. Many P1B-type ATPases, but not all of them, also contain a diverse arrangement of cytoplasmic metal binding domains (MBDs). In spite of their structural differences, all N- and C-terminal MBDs appear to control the enzyme turnover rate without affecting metal binding to transmembrane transport sites. In addition, eukaryotic Cu+-ATPases have multiple N-MBD regions that participate in the metal dependent targeting and localization of these proteins. The current knowledge of structure-function relationships among the different P1B-ATPases allows for a description of selectivity, regulation and transport mechanisms. Moreover, it provides a framework to understand mutations in human Cu+-ATPases (ATP7A and ATP7B) that lead to Menkes and Wilson diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Defining features of P1B-ATPases
P1B-ATPasesFootnote 1 are a subgroup of P-ATPases that transport heavy metals (Cu+, Cu2+, Zn2+, Co2+) across biological membranes (Lutsenko and Kaplan 1995; Solioz and Vulpe 1996; Axelsen and Palmgren 1998; Rensing et al. 1999; Argüello 2003; Williams and Mills 2005). An immediate consequence of their metal specificity and the chemical similarities among transition metals is that these pumps transport alternative non-physiological substrates; for instance, Cu+-ATPases transport Ag+ while Zn2+-ATPases can transport Cd2+ and Pb2+. P1B-ATPases share a common catalytic mechanism with the archetypical P-ATPases (Na+, K+-ATPase, Ca2+-ATPase, H+-ATPase) (Fig. 1). This mechanism couples the catalytic phosphorylation of Asp in the invariant DKTGT sequence to the transmembrane translocation of the metals. P1B-ATPases have structural features such as metal-binding signature sequences in their transmembrane segments (TM), topological arrangements with eight or six TMs, and regulatory cytoplasmic metal binding domains (MDB), that distinguish them from their P-ATPase counterparts (Fig. 2). Consistent with their substrate specificities and direction of transport, i.e., metal efflux from the cytoplasm, they confer metal tolerance to archaea and bacteria, while in higher eukaryotes they are responsible for metal micronutrient absorption, distribution and clearance.

P1B-ATPases catalytic cycle. E1, E2, E1P and E2P represent the basic conformations that the enzyme can assume. Mn+ represents a metal transported by these enzymes. n indicates the uncertainty on the specific stoichiometry of transport. Mn+ cyt and Mn+ out represents the cytoplasmic or extracellular/luminal localization of the transported metal

Schematic illustration of the topology and main domains present in P1B-ATPases. Transmembrane segments, H1 to H8, are indicated. The relative locations of the cytoplasmic actuator (A), phosphorylation (P) and nucleotide (N) domains are shown. The conserved amino acids in H6, H7 and H8 forming the transmembrane metal binding sites (TM-MBS) are symbolized by blue dots. The presence of N- and C-terminal metal binding domains (MBDs) with diverse structures is represented by yellow and red rectangles
Distribution and physiological roles
Different from other P-ATPases subfamilies, P1B-ATPases are found in all life kingdoms, from extremophilic archaea to man (Fig. 3). P1B-ATPases are present in most reported archaeal genomes (Argüello 2003; De Hertogh et al. 2004). Archaeal P1B-ATPases present a variety of substrate specificities and considerable structural stability (Mandal et al. 2002; Argüello 2003; Mana-Capelli et al. 2003; Baker-Austin et al. 2005) (Cattoni D, González-Flecha, FL, Argüello JM, unpublished results). These properties are linked to the extremophilic character of the hosting organisms. For instance, increased Cu+-ATPase transcript levels have been observed in Ferroplasma acidarmanus, an organism that tolerates copper at levels of 20 g/l (Baker-Austin et al. 2005). Alternatively, the chemolithoautotroph Archaeoglobus fulgidus has two P1B-ATPases, CopA and CopB, able to transport Cu+ and Cu2+ respectively, suggesting the need to extrude alternative copper forms depending on the organism redox status (Mandal et al. 2002; Mana-Capelli et al. 2003). In view of their stability and diversity, these proteins appear as a relevant resource to explore structural-functional characteristics and biotechnological applications.

Phylogenetic tree of the P1B-type ATPases. The tree was prepared from a Clustal W alignment of representative sequences of P1B-type ATPases. The relative abundance of sequences from each subgroup has been maintained. The metal specificity and the structural characteristics are indicated next to the subgroup denomination. Amino acids in TMs proposed to participate in determining metal selectivity. Black blocks represent His-rich N-MBDs; orange blocks, CXXC N-MBDs; and red, His and Cys rich N- and C-MBDs
P1B-ATPases were initially identified in prokaryotic organisms, like Staphylococcus aureous plasmid pI258 (Nucifora et al. 1989), Rhizobium meliloti (Kahn et al. 1989) and Enterococcus hirae (Odermatt et al. 1993). In these organisms, their function is to maintain metal quotas, specially those of copper and zinc. This role has been demonstrated by gene knockout resulting in high sensitivity to the metal transported by the encoded protein. This experimental approach, together with functional complementation, has served to identify various P1B-ATPases and their multiple substrates (Odermatt et al. 1993; Phung et al. 1994; Rensing et al. 1997; Rutherford et al. 1999; Rensing et al. 2000; Tottey et al. 2001). It is interesting that some prokaryotes (Mycobacterium tuberculosis, Lactococcus lactis, Pseudomonas aeruginosa, etc.) have two or more P1B-ATPases of similar metal specificity (Argüello 2003). The reasons for this abundance, and apparent redundancy, have not been explored.
Genomes of fungi and animals contain two genes coding for Cu+-ATPases (Axelsen and Palmgren 1998; Argüello 2003). Among these, the better characterized are the two present in humans, ATP7A and ATP7B. Mutations in these genes lead to Menkes syndrome and Wilson disease respectively (Bull et al. 1993; Vulpe et al. 1993; Bull and Cox 1994; Lutsenko et al. 2003). Wilson disease is characterized by high copper levels in the liver, blood and brain, with consequent neurologic and hepatic disorders. Mutations in the Menkes ATPase (MNKP) lead to poor copper uptake from the intestine resulting in neurodegeneration and connective tissue abnormalities. These symptoms are associated with the particular subcellular distribution and specific organ expression of these enzymes. MNKP is expressed in the intestine, blood brain barrier and several other organs (Vulpe et al. 1993). In contrast, Wilson disease protein (WNDP) expression is predominant in the liver. Both enzymes localize to the trans-Golgi network and undergo Cu-dependent trafficking (Petris et al. 1996; Hung et al. 1997). However, while the MNKP is relocated to the cell plasma membrane in various tissues (Petris et al. 1996), the WNDP is targeted to vesicles proximal to the plasma membrane of liver canicular cells (Forbes et al. 1999; Schaefer et al. 1999).
Plants differ significantly from other organisms in the number and selectivity of their P1B-ATPases (Williams and Mills 2005). For instance, Arabidopsis thaliana contains four Cu+-ATPases (HMA5-8) and three Zn2+-ATPases (HMA2-4). HMA1 has been reported to be a Cu+-ATPase although its transmembrane metal binding site (TM-MBS) appears to be different from that of Cu+-transporting ATPases (Seigneurin-Berny et al. 2006). Plant Cu+-ATPases are responsible for Cu+ transport into a post-Golgi compartment (HMA7) (Woeste and Kieber 2000) and the chloroplast (HMA6, HMA8 and possibly HMA1) (Abdel-Ghany et al. 2005; Seigneurin-Berny et al. 2006). HMA5 appears to be involved in copper detoxification in roots (Andrés-Colas et al. 2006). The Zn2+-ATPases HMA2 and HMA4 are located in the plasma membrane and expressed primarily in the vasculature of shoots and roots (Hussain et al. 2004). hma2 knockout plants present high Zn2+ levels (Eren and Argüello 2004), while the hma4 knockout plants show reduced Zn2+ levels (Hussain et al. 2004). Along this line, over-expression of HMA4 increases root-to-shoot Zn2+ translocation (Verret et al. 2004). Considering their distribution, these observations suggest that HMA2 and HMA4 participate in Zn2+ loading into the phloem and xylem respectively.
The catalytic mechanism
P1B-ATPases transport metals across membranes following the classical E1/E2 Albers-Post catalytical cycle (Fig. 1). This transport mechanism has been studied in detail in P2-ATPases (Na+,K+-, Ca2+-, and H+,K+-ATPases) (MacLennan et al. 1997; Kaplan 2002). Catalytic enzyme phosphorylation occurs upon ATP binding with high (μM) affinity to the ATP binding domain (ATP-BD) and metal binding to the TM-MBS from the cytoplasmic side (Steps 4 and 1). Upon phosphorylation, the metal is likely occluded within the TM region and not accessible from the aqueous media. The subsequent conformational change allows for metal deoclussion, release to the extracellular (vesicular/luminal) compartment, and subsequent enzyme dephosphorylation (Step 3). The enzyme then returns to the E1 conformation upon ATP binding to the E2 form with low (mM) affinity (Step 4).
Biochemical studies have corroborated this model for various P1B-ATPases while explaining aspects that are unique to heavy metal transport. Heterologously expressed eukaryote Cu+-ATPases have served to characterize transport into sealed vesicles, phosphorylation and dephosphorylation reactions in isolated proteins, and metal transfer among Cu-chaperones and N-terminal metal binding domains (MBDs) (Voskoboinik et al. 1998; Huffman and O’Halloran 2000; Wernimont et al. 2000; Tsivkovskii et al. 2002; Walker et al. 2002). ATPase activity, transport, direct metal binding to the TM-MBS, and phosphorylation reactions have been measured in eukaryote Zn2+-ATPases and various prokaryote and archaeal enzymes, either in isolated membranes or solubilized pure enzyme preparations (Sharma et al. 2000; Fan and Rosen 2002; Mandal et al. 2002; Mana-Capelli et al. 2003; Mandal and Argüello 2003; Eren and Argüello 2004; Okkeri et al. 2004; Liu et al. 2006). These studies have allowed the characterization of mechanistic requirements, selectivity, and regulatory aspects independently of in vivo constraints associated with targeting, protein interactions, and interlinked homeostasis mechanisms of non-metal substrates.
Direction of transport and stoichiometry
Transport experiments indicate that P1B-ATPases drive metal efflux from the cytoplasmic compartment (Rensing et al. 1997; Voskoboinik et al. 1998; Fan and Rosen 2002; Mana-Capelli et al. 2003; Eren and Argüello 2004). This is in agreement with a catalytic mechanism where the E1 form of the enzyme (TM-MBS open to the cytoplasmic side) binds the transported metal and ATP (Fig. 1). Early reports have suggested that some Cu+-ATPases, although structurally homologous to well-characterized enzymes, might drive metal influx into the cytoplasm (Odermatt et al. 1993; Tottey et al. 2001). However, the influx of metal would require binding of another transport substrate (proton?) to the E1 form to allow the coupled ATP hydrolysis and enzyme phosphorylation, with the inwardly transported metal driving enzyme dephosphorylation. Further experiments would be required to test this alternative mechanism.
The coupling of solute transport with ATP hydrolysis in a specific stoichiometry is a requirement of the E1/E2 mechanisms (MacLennan et al. 1997; Kaplan 2002). Because of experimental difficulties to obtain highly active preparations of sealed everted vesicles or, alternatively, conditions to stabilize the metal occluded (E1P(metal)) forms, these parameters have not been established for most P1B-ATPases. However, Mitra and collaborators recently showed binding of 1 Zn2+ per ATPase to the TM-MBS of E. coli ZntA in the absence of other enzyme substrates (Liu et al. 2006). Although it could be argued whether Zn2+ TM-MBS were fully occupied, this relevant data is the first showing isolated metal binding to the TM-MBS.
The membrane topology of P1B-ATPases
When compared to other P-ATPases, P1B-ATPases have a distinct structure characterized by a reduced number of TMs, smaller ATP binding domains (ATP-BD), and the presence of N- or C-terminal MBDs in many of them (Fig. 2). The presence of eight TMs has been experimentally established in two bacterial enzymes (Melchers et al. 1996; Tsai et al. 2002). A similar topology emerges through computational analysis of most P1B-ATPases (Fig. 3). However, a subgroup of these enzymes appears to have only six TMs. This particular topology is observed in one of the major P1B-ATPases subgroups, P1B-4, and in a number of isolated proteins lacking signature sequences that define Cu+-, Zn2+- or Cu2+-ATPases (subgroup P1B-5). The six TM topology is supported by single and multiple sequence analysis. Furthermore, while retaining the central cytoplasmic regions necessary for ATP binding and hydrolysis, these proteins are relatively small with short N- and C-terminal ends; thus, they contain no possible sequence to form the two additional segments observed in the P1B-1, P1B-2 and P1B-3 subgroups (Fig. 3). The described topologies differentiate P1B-ATPases from other subfamilies that contain ten (P2 and P3) or twelve (P4 and P5) TMs (Lutsenko and Kaplan 1995; Axelsen and Palmgren 1998). P1B-ATPases also present a particular distribution of TMs with respect to the large cytoplasmic loop forming the ATP-BD. While P1B-ATPases have two TMs on the C-terminal end of the ATP-BD, other P-ATPases have either four (P1A) or six (P2, P3, P4 and P5).
In spite of the indicated differences, a common pattern is evident among these heavy metal ATPases. They all have a core structure formed by the five TMs flanking the two largest cytoplasmic loops (Fig. 3). These central components appear to confer their basic functionality to these enzymes, i.e., the capability to transport metals using the energy resulting from ATP hydrolysis.
The transmembrane metal binding site
The transmembrane metal binding sites (TM-MBS) of P1B-ATPases are responsible for metal recognition and movement across the membrane permeability barrier. While there is a significant wealth of information on metal coordination in proteins where metals play structural and catalytic roles, little is known about the metal sites of transporters that bind and release the metals with high turnover (approximately 200 min- in the case of P1B-ATPases). For instance, the requirement of conserved transmembrane His, Glu, Asp, Ser and Met residues for the metal translocation by various transporters has been established, but a detailed description of the arrangement of these metal binding ligands is still lacking. Analysis of the first available sequences indicated that P1B-ATPases have a CPC sequence in the center of their sixth TM (H6), with a few exceptions containing CPH (Bull and Cox 1994; Solioz and Vulpe 1996). While the Pro is conserved in all P-ATPases, the CPX motif appeared as a defining element of these enzymes that likely participate in metal coordination during transport. Mutations of these Cys impair enzyme function (Yoshimizu et al. 1998; Fan and Rosen 2002; Mandal and Argüello 2003; Lowe et al. 2004). These mutants seemed unable to bind Cu+ but capable of interacting with nucleotides and undergoing basic conformational changes (Mandal and Argüello 2003). Although the CPC sequences are the most frequently observed, a current analysis of the available genomes reveals the presence of alternative sequences (SPC, CPS, CPT, CPA, CPG, CPD) in putative P1B-ATPases. These proteins contain the DKTGT signature sequence and the characteristic topology of these enzymes. This observation highlights the diversity of these pumps and suggests the possibility of metal specificities yet to be identified.
Early studies of the Ca2+- and Na+,K+-ATPases (MacLennan et al. 1997; Lingrel et al. 1998) and more recently the evidence provided by the structure of the Ca2+-ATPase SERCA1 (Toyoshima and Inesi 2004), indicate that their transmembrane cation binding sites are constituted by invariant polar side chains located in the three TMs flanking the ATP-BD (one in the N-terminal side and two on the C-terminal end). Following a similar pattern, conserved amino acids were identified in the equivalent TMs of P1B-ATPases (Argüello 2003). The observed signature sequences pointed out the various TM-MBS present in these proteins and allowed for the classification of proteins into subgroups with distinct metal binding and transport specificities (Fig. 3).
Subgroup P1B-1 comprises all Cu+-ATPases, including the human WNDP and MNKP. In the center of their transmembrane region these proteins contain conserved residues Asn, Tyr in H7, Met and Ser in H8, and the CPC sequence in H6 (Argüello 2003). The functional roles of these residues were tested by using as a model the hyperthermophilic CopA from A. fulgidus (Mandal et al. 2004). CopA carrying mutations in the mentioned residues were inactive. They were phosphorylated by inorganic phosphate (Pi) (Step 3, Fig. 1) showing that they retain their overall structure and could undergo major conformational transitions. However, mutant AfCopAs were not phosphorylated by ATP in the presence of Cu+ (Step 1, Fig. 1) nor was Cu+ able to prevent enzyme phosphorylation by Pi (Step 4, Fig. 1). Interestingly, a mutation of the conserved Ser in H8 of WNDP has been identified in some Wilson disease patients (Loudianos et al. 1999). Mutations in the immediate vicinity of these residues or in other TMs have no significant effects in enzyme activity or in Cu+ dependent partial reactions (Lowe et al. 2004; Mandal et al. 2004). These observations suggest the participation of specific invariant residues (two Cys, Asn, Tyr, Met and Ser) in Cu+ transport by P1B-ATPases. The identification of these side chains provides a first view of the putative TM-MBS for Cu+-ATPases. However, further information is necessary to understand Cu+ binding to the TM-MBS and subsequent transport. For instance, the stoichiometry of transport and the direct participation of these residues in initial ion binding, occlusion, or release remain to be established. In this direction, it seems likely that the second Cys in H6, proximal to the cytoplasmic side, would be required for metal binding, while the first Cys would be necessary for Cu+ dissociation or enzyme dephosphorylation (Lowe et al. 2004).
The P1B-2 group includes the Zn2+-ATPases. These enzymes contain the CPC in H6, a Lys in H7 and Asp and Gly in H8, as putative components of the corresponding TM-MBS (Fig. 3). Although the roles of the Lys and Gly have not been explored, mutations of Asp714 of Escherichia coli ZntA yields inactive enzymes (Dutta et al. 2006). Interestingly, Asp714His and Asp714Glu mutants were able to bind Zn2+ and undergo phosphorylation in the presence of Pi (in the absence of metal). However, they could not be phosphorylated by ATP in the presence of Zn2+. These results further support the concept that TMs H6, H7 and H8 contribute amino acids to the TM-MBS.
Group P1B-3 corresponds to the Cu2+-ATPases. In these proteins, the second Cys in H6 is replaced by His while amino acids in H7 and H8 are similar to those in the Cu+-ATPase subgroup (Fig. 3). Taking into account the presence of the imidazolium side chain (a hard Lewis base), the preferences of these enzymes to transport Cu2+ (an intermediate Lewis base) rather than Cu+, is not surprising (Argüello 2003; Mana-Capelli et al. 2003). Although the lack of activity in E. hirae CopB carrying a → SPH mutation has been reported (Bissig et al. 2001), no further studies have been performed on the TM-MBS of P1B-3 proteins. Similarly, the functional roles of amino acids conserved among proteins in subgroup P1B-4 have not been explored. Moreover, the metal specificity of these ATPases remains unclear. Studies of Synechocystis PCC6803 CoaT have shown that a knockout of the encoding gene leads to increased levels of intracellular Co2+ and reduced Co2+ tolerance (Rutherford et al. 1999). A recent study characterized A. thaliana HMA1, an enzyme that localizes in the chloroplast envelope and contains the signature amino acids and the topology that differentiate the P1B-4-ATPases. The hma1 knockout showed high copper content in the chloroplast and a phenotype compatible with this alteration. Enzymatic determinations using chloroplast membranes from plants over-expressing HMA1 showed higher levels of ATP hydrolysis in the presence of 3 mM copper. No activation was observed in the presence of 3 mM Co2+, Zn2+, Mn2+ or Fe2+. Considering both studies, it is difficult to delineate the metal specificity of P1B-4-ATPases and further characterization of other members of this subgroup seems necessary.
The central role of TM-MBDs for determining the specificity of P1B-ATPases is also supported by experiments looking at the metal selectivity of enzymes lacking the N- and C-MBDs. The relative selectivity, both in terms of V max and metal K1/2 for activation, among the various transported metals (Cu+/Ag+, Cu2+/Cu+/Ag+ or Zn2+, Cd2+/Pb2+) is maintained when the N-MBDs are removed or mutated (Mitra and Sharma 2001; Mana-Capelli et al. 2003; Mandal and Argüello 2003). Furthermore, E. coli ZntA chimeras containing N-MBDs from the WNDP Cu+-ATPase retain Zn2+-ATPase activity (Hou et al. 2001).
Metal binding to the TM-MBS
Enzyme phosphorylation by ATP, subsequent turnover and transport requires metal binding to the TM-MBS. This is independent of metal binding to the N- and C-MBDs, since truncated ATPases lacking these domains or proteins carrying mutations yielding N-MBDs unable to bind metals are phosphorylated and transport metals (Voskoboinik et al. 1999; Bal et al. 2001; Mitra and Sharma 2001; Fan and Rosen 2002; Mana-Capelli et al. 2003; Mandal and Argüello 2003). The case of PINA (Pineal Night-Specific ATPase), a splice variant of ATP7B gene lacking the WDNP six N-MBDs, is an example of truncated proteins in vivo functionality (Borjigin et al. 1999).
Considering the metal-protein interaction, it should be kept in mind the low in vivo free Cu+ and Zn2+ concentrations associated with the presence of specific chaperone proteins and/or numerous ligands (Outten and O’Halloran 2001; Changela et al. 2003). Metals activate P1B-ATPases with apparent affinities (K 1/2) in the 0.1-3 μM range (Okkeri and Haltia 1999; Voskoboinik et al. 1999; Sharma et al. 2000; Fan and Rosen 2002; Mandal et al. 2002; Tsivkovskii et al. 2002; Mana-Capelli et al. 2003; Eren and Argüello 2004). Most of these assays have been performed in the presence of various metal ligands such as DTT, Cys, and of course, ATP (Sharma et al. 2000; Fan and Rosen 2002; Mandal et al. 2002; Eren and Argüello 2004). Moreover, WNDP phosphorylation by ATP is activated by the Cu-Atox1 complex with a 2 μM K1/2 (Walker et al. 2002). Therefore, these K 1/2 values do not refer to free metal concentrations but to the total metal in the media. Considering the low dissociation constants for the soluble metal complexes (metal-thiolate, metal-chaperone, metal-ATP) (Martell and Smith 2004), it can be proposed that these complexes deliver the metal directly to the TM-MBS, perhaps by a kinetically controlled ligand exchange. This mechanism was proposed to explain the metal transfer between copper chaperones and N-MBDs (Huffman and O’Halloran 2000). The question remains whether this is a plausible in vivo mechanism of metal delivery to the TM-MBS. This is particularly relevant in the case of Cu+-ATPases where cytoplasmic Cu+ is bound to chaperones. Direct metal transfer from the Cu-chaperones to the TM-MBS (or from the N-MBDs to the TM-MBS) has not been shown. However, Lutsenko and her collaborators have shown that WNDP phosphorylation by ATP in the presence of Cu-Atox1, requires the second N-MBD (Walker et al. 2004). This finding suggests that at least in proteins with multiple N-MBDs, like WNDP or MNKP, Cu+ might be delivered to the TM-MBS by the N-MBDs. Considering this result and the presence of enzymes lacking N-MBDs; for instance, the PINA variant and the members of subgroup P1B-4, alternative mechanisms with direct cytoplasmic metal delivery to the TM-MBS might be expected to emerge. Independent of metal delivery to TM-MBS via a cytoplasmic MBD or directly to TM-MBDs by a chaperone, the feasibility of ligand exchange with a transmembrane binding site formed by residues from H6, H7, and H8 can be examined by comparison with the structurally related SERCA1. The structure of this Ca2+-ATPase shows that Ca2+ access to the transmembrane binding sites is through a relatively wide (8-10Å) funnel that contains one the Ca2+ ligands (equivalent to the proximal Cys in P1B-ATPases H6) at its bottom (Toyoshima and Nomura 2002). Then, among other possibilities, it could be hypothesized that if a similar funnel were present in P1B-ATPases, ligand-bound metals might be “delivered” directly into the TM-MBS.
The actuator and ATP binding domains
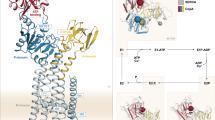
The large cytoplasmic loop between H6 and H7 of P1B-ATPases contains the DKTGT sequence conserved in all P-ATPases (Fig. 2). The loop, referred to as ATP-BD, encompasses the nucleotide binding (N) and the phosphorylation (P) domains. The smaller loop between H4 and H5 forms the actuator (A) domain. Recently the structures of the isolated ATP-BD and A-domain of AfCopA and the N-domain of WNDP have been reported (Dmitriev et al. 2006; Sazinsky et al. 2006a, b) (Fig. 4). The A-domain of AfCopA shows a 10 β-strand core with two α-helices connecting with the TMs present in the full-length protein (Fig. 4A) (Sazinsky et al. 2006a). In spite of their small sequence homology (26%), the folding of AfCopA is highly similar to that of the A-domain of SERCA1, the P2-type Ca2+-ATPase (Toyoshima et al. 2000; Toyoshima and Inesi 2004; Toyoshima et al. 2004). In particular, the presence of the highly conserved (S/T)GE(P/S) sequence at the tip of a solvent accessible loop on the side of the A-domain appears similar. This has relevant functional implications. In the Ca2+-ATPase, the interactions of this loop with the P-domain during enzyme phosphorylation/dephosphorylation appears critical since they drive the rotation of the A-domain with a subsequent rearrangement of TMs (Toyoshima and Nomura 2002; Olesen et al. 2004; Toyoshima and Inesi 2004; Toyoshima et al. 2004). This rearrangement, in turn, leads to metal deocclusion and release (Step 2, Fig. 1). Although the different disposition of TMs in the P1B-ATPases might require different transmembrane movements, the structural similarities suggest an equivalent gating mechanism for metal release.

Structure of the A-, P- and N-domains of Archaeoglobus fulgidus CopA. Structures in A, B, D and E are presented with the segments that would be proximal to the TMs in the full-protein on the top of the models. A, AfCopA A-domain. Location of conserved SGEP sequence is shown. B, AfCopA ATP-BD. P-domain and N-domain are labeled. The location of conserved D424 is indicated. An AMP is modeled in the nucleotide binding site. Surfaces of conserved residues predicted to bind AMP are colored by atom type with oxygen red, carbon grey. C, Superimposed AfCopA (red) and WNDP (grey) N-domains. A flexible loop between A1114 and T1143 in the WNDP was removed from the model for clarity. D, Mapping of WNDP and MNKP mutations in the structure of AfCopA A-domain. E, Mapping of WNDP and MNKP mutations in the structure of AfCopA ATP-BD
The ATP-BD structure shows the P- and N-domains joined by two short loops (the hinge region) (Sazinsky et al. 2006b). The P-domain consists of six-stranded parallel β-sheets sandwiched between three short α-helices (Fig. 4B). This domain contains the DKTGT sequence and a number of residues conserved in all P-ATPases that interact with the ATP γ-phosphate during binding and hydrolysis (Sørensen et al. 2004). Although the AfCopA P-domain is smaller and lacks some of the solvent exposed helices present in SERCA1 (Toyoshima et al. 2000) both proteins have a similar global fold.
The N-domain of both, WNDP and AfCopA, share a similar structure in accordance with their strong homology (Fig. 4C) (Dmitriev et al. 2006; Sazinsky et al. 2006b). The N-domains consist of six antiparallel β-sheets flanked by four α-helices. A significant difference is a 29 amino acid flexible loop present in the WNDP (Dmitriev et al. 2006) but not in AfCopA. This fragment might be associated with roles peculiar to the eukaryote enzymes including perhaps alternative regulatory mechanisms and required targeting. Although these N-domains have little homology with that of SERCA1 (<25%), they reveal the same basic structure just lacking several 7-10 amino acid insertions located in the periphery of the SERCA1 structure (Toyoshima et al. 2000). More importantly, the amino acids forming the ATP binding site are distinct for the P1B- and P2-ATPases. P1B-ATPases have a stronger similarity with the homologous region of KdpB, a P1A-ATPase (Haupt et al. 2004; Dmitriev et al. 2006; Sazinsky et al. 2006a, b). The structures of KdpB and WNDP N-domains have been determined in the presence of nucleotides. Analysis of residues participating in nucleotide binding (His1069, Gly1099, Gly1101, Gly1149 and Asn1150 in the WNDP) shows that P1A- and P1B-ATPases have a unique ATP binding site distinct from that in P2-ATPases. The involvement of some of these amino acids in ATP binding is supported by mutagenesis studies in the WNDP (Tsivkovskii et al. 2003; Morgan et al. 2004), E. coli ZntA (Okkeri and Haltia 1999; Okkeri et al. 2004), and E. hirae CopB (Bissig et al. 2001).
The structural description of the A, P, and N domains support the concept that, in spite of the differences among the sequences of the various P-ATPase subfamilies, they employ similar structures, not only to bind their ligands, but to implement the critical conformation changes required for energy transduction. Furthermore, these structures provide a framework to interpret the likely effect of many of the mutations leading to Wilson and Menkes diseases occurring in regions that are conserved in AfCopA. We have mapped the position of WNDP and MNKP mutations within the structures of the AfCopA ATP-BD and A-domain (Fig. 4D and E) (Cox and Moore 2002; Sazinsky et al. 2006a, b). This analysis allows differentiating likely effects on different enzyme functions. Mutations at positions highlighted in red in the ATP-BD likely affect enzyme phosphorylation, while the red positions in the A domain indicted those that might alter interactions among the A- and P-domains. Those in yellow might affect the overall enzyme stability rather than a specific ligand interaction. Blue marks positions in the N-domain that when mutated might affect ATP binding, among these, His1069 in the WNDP. In the A-domain, mutations highlighted in red might affect the interactions of the A- and P-domain. Finally, in green are positions located in an accessible region of the A-domain that is opposite to the loop interacting with the P-domain. Considering their location these mutations might affect intra- or inter-molecular domain-domain interactions. Since rotation of the A-domain controls the rate limiting metal deoclussion (Step 3, Fig. 1), and a regulatory for role controlling enzyme turnover has been suggested for the cytoplasmic MBDs (see below), it is tempting to hypothesize that these mutations might point out the sites of a functionally relevant N-MBD-A-domain interaction.
The cytoplasmic metal binding domains
Most P1B-ATPases have various types of cytoplasmic metal binding domains (MBD) located either in the N-terminus (N-MBD) or C-terminus (C-MBD) (Figs. 2 and 3) (Tables 1 and 2). The N-MBDs observed in Cu+-ATPases and some bacterial Zn2+-ATPases are 60-70 amino acids domains with a highly conserved CXXC metal-binding sequence (Table 1) (Rensing et al. 1999; Arnesano et al. 2002; Lutsenko et al. 2003). Eukaryote Cu+-ATPases contain multiple repeats of this domain. The human WNDP and MNKP have six N-MBDs that have been the focus of many studies (Arnesano et al. 2002; Lutsenko et al. 2003). Alternatively, archaea and prokaryote enzymes have usually one-two N-MBDs. The high-resolution structures of several of these domains in their apo and metal-bound forms have been reported (Gitschier et al. 1998; Banci et al. 2001; Banci et al. 2002). They present a βαββαβ fold similar to the well-described Cu-chaperones like human Atox1, yeast Atx1, and prokaryote CopZ (Rosenzweig et al. 1999; Wimmer et al. 1999; Arnesano et al. 2002). In vitro, these N-MBDs can bind both monovalent and divalent cations including Cu+, Cu2+, Zn2+ and Cd2+ (DiDonato et al. 1997; Lutsenko et al. 1997; Jensen et al. 1999; Liu et al. 2005). In vivo, N-MBDs receive Cu+ from the corresponding Cu-chaperones (Hamza et al. 1999; Larin et al. 1999; Huffman and O’Halloran 2000; Wernimont et al. 2000). Their metal selectivity appears to be determined by the specific N-MBD-chaperone interaction via electrostatic and hydrophobic interactions that align the metal binding sites for rapid ligand exchange (Wernimont et al. 2000; Arnesano et al. 2002). This metal exchange has not been established for Zn2+-ATPases carrying CXXC containing N-MBDs since no Zn-chaperone (or equivalent molecule) has been identified. Recent structural analysis of E. coli ZntA N-MBD, suggest that Asp in the DCXXC sequence might provide the electrostatic requirement to confer Zn2+ specificity to these domains (Banci et al. 2002).
Some prokaryote Zn2+-ATPases have His-rich N-MBDs [(HX) n (n = 2-6)] alone or together with the CXXC N-MBDs (Table 1). Similar sequences have been observed in ZIP and Cation Diffusion Facilitator (CDF) families, located in cytoplasmic loops joining TMs (Paulsen and Saier 1997; Eng et al. 1998). Cu2+-ATPases (subgroup PIB-3) and a few members of subgroup PIB-4 also have His rich N-MBDs. However, these are His stretches instead of HX repeats. Although it is clear that these have functional roles (Mana-Capelli et al. 2003), the metal binding specificity and properties of these domains have not been characterized.
All plant Zn2+-ATPases contain unique sequences in both the N-terminus and C-terminus. Plant N-MBDs appear homologous to those described for Cu+-ATPases and prokaryote Zn2+-ATPases. However, the CXXC sequences are replaced by CCXSE (x = S, T, P) (Table 1). These changes appear critical for selective Zn2+ binding since A. thaliana HMA2 N-MBD binds Zn2+ in vitro but not Cu+ (Eren and Argüello, unpublished results). Plant Zn2+-ATPases also have unusually long C-termini containing numerous His and Cys (Table 2). These have various lengths and present two different patterns. The putative C-MBD from A. thaliana HMA3 and its homologs, have numerous Cys but no (or few) His residues. Alternatively, HMA2, HMA4 and homologous proteins have His- and Cys-rich C-MBD. We have characterized metal binding to the HMA2 C-MBD. This domain binds three Zn2+ (or Cd2+) with an apparent K d in the nanomolar range. One of the Zn2+ binding sites is apparently formed by one Cys and three His, while the others are constituted by four His (Eren and Argüello, unpublished results).
While the various structures observed for N-MBDs and C-MBDs add to the complexity of P1B-ATPases, these appear as different strategies to accomplish similar functions related to the regulation of these pumps.
Regulatory roles of cytoplasmic MBDs
MBDs are not essential for ATP hydrolysis and metal transport by P1B-ATPases (Voskoboinik et al. 1999; Bal et al. 2001; Mitra and Sharma 2001; Fan and Rosen 2002; Mana-Capelli et al. 2003; Mandal and Argüello 2003). Numerous studies have pointed out alternative regulatory roles for these domains (Rensing et al. 1999; Fatemi and Sarkar 2002; Lutsenko et al. 2003). They are required for the Cu-dependent targeting of MNKD and WNDP (Petris et al. 1996; Forbes et al. 1999; Schaefer et al. 1999). However, these critical physiological phenomena are beyond the scope of this discussion. Alternatively, it has also been observed that the N-MBDs control the rate of P1B-ATPases turnover with little or no change in metal affinity (Voskoboinik et al. 1999; Mitra and Sharma 2001; Voskoboinik et al. 2001; Fan and Rosen 2002; Mana-Capelli et al. 2003; Mandal and Argüello 2003). These findings suggested that metal binding to N-MBDs affects the rate-limiting step of these enzymes catalytic cycle. Studies using A. fulgidus CopA and CopB as models confirmed that the rate limiting conformational change associated with metal release/dephosphorylation is slower when the N-NMBs are removed or made unable to bind metals by specific mutations (Mana-Capelli et al. 2003; Mandal and Argüello 2003). This effect is likely achieved by Cu-dependent changes in the interactions of N-MBDs with other protein domains. Supporting this idea, the Cu+ dependent interaction of Wilson disease protein N-MBDs with the large ATP binding cytoplasmic loop has been shown (Tsivkovskii et al. 2001).
Summary and future directions
Significant advances have been produced in the understanding of the heavy metal transport mechanisms by P1B-ATPases. The structures of domains responsible for ATP binding and hydrolysis, and energy transduction have been determined. The presence of the transmembrane metal binding site constituted by side chains from residues in TMs H6, H7 and H8, has been established. This has been an important step toward understanding the mechanisms of selectivity and metal translocation across the membrane. Understanding the catalytic cycle has facilitated probing the direction of transport and the regulatory role of cytoplasmic metal binding domains. Future molecular studies will likely address the detailed structure of the transmembrane region, the mechanisms of metal delivery to transmembrane metal binding site, the interactions among cytoplasmic domains and their functional consequences.
Notes
For simplicity P-type ATPases will be referred as P-ATPases, P1B-ATPase, etc.
References
Abdel-Ghany SE, Muller-Moule P, Niyogi KK et al (2005) Two P-type ATPases are required for copper delivery in Arabidopsis thaliana chloroplasts. Plant Cell 17:1233–1251
Andrés-Colas N, Sancenon V, Rodríguez-Navarro S et al (2006) The Arabidopsis heavy metal P-type ATPase HMA5 interacts with metallochaperones and functions in copper detoxification of roots. Plant J 45:225–236
Argüello JM (2003) Identification of ion selectivity determinants in heavy metal transport P1B-type ATPases. J Membr Biol 195:93–108
Arnesano F, Banci L, Bertini I et al (2002) Metallochaperones and metal-transporting ATPases: a comparative analysis of sequences and structures. Genome Res 12:255–271
Axelsen KB, Palmgren MG (1998) Evolution of substrate specificities in the P-type ATPase superfamily. J Mol Evol 46:84–101
Baker-Austin C, Dopson M, Wexler M et al (2005) Molecular insight into extreme copper resistance in the extremophilic archaeon Ferroplasma acidarmanus Fer1. Microbiology 151:2637–2646
Bal N, Mintz E, Guillain F et al (2001) A possible regulatory role for the metal-binding domain of CadA, the Listeria monocytogenes Cd2+-ATPase. FEBS Lett 506:249–252
Banci L, Bertini I, Ciofi-Baffoni S et al (2002) A new Zinc-protein coordination site in intracellular metal trafficking: Solution structure of the apo and Zn(II) forms of ZntA(46–118). J Mol Biol 323:883–897
Banci L, Bertini I, Ciofi-Baffoni S et al (2001) Solution structure of the yeast copper transporter domain Ccc2a in the apo and Cu(I)-loaded states. J Biol Chem 276:8415–8426
Bissig KD, Wunderli-Ye H, Duda PW et al (2001) Structure-function analysis of purified Enterococcus hirae CopB copper ATPase: effect of Menkes/Wilson disease mutation homologues. Biochem J 357:217–223
Borjigin J, Payne AS, Deng J et al (1999) A novel pineal night-specific ATPase encoded by the Wilson disease gene. J Neurosci 19:1018–1026
Bull PC, Cox DW (1994) Wilson disease and Menkes disease: new handles on heavy-metal transport. Trends Genet 10:246–252
Bull PC, Thomas GR, Rommens JM et al (1993) The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet 5:327–337
Changela A, Chen K, Xue Y et al (2003) Molecular basis of metal-ion selectivity and zeptomolar sensitivity by CueR. Science 301:1383–1387
Cox DW, Moore SD (2002) Copper transporting P-Type ATPases and human disease. J Bioenerg Biomembr 34:333–338
De Hertogh B, Lantin AC, Baret PV et al (2004) The archaeal P-type ATPases. J Bioenerg Biomembr 36:135–142
DiDonato M, Narindrasorasak S, Forbes JR et al (1997) Expression, purification, and metal binding properties of the N-terminal domain from the Wilson disease putative copper-transporting ATPase (ATP7B). J Biol Chem 272:33279–33282
Dmitriev O, Tsivkovskii R, Abildgaard F et al (2006) Solution structure of the N-domain of Wilson disease protein: distinct nucleotide-binding environment and effects of disease mutations. Proc Natl Acad Sci USA 103:5302–5307
Dutta SJ, Liu J, Hou Z et al (2006) Conserved aspartic acid 714 in transmembrane segment 8 of the ZntA subgroup of P1B-type ATPases is a metal-binding residue. Biochemistry 45:5923–5931
Eng BH, Guerinot ML, Eide D et al (1998) Sequence analyses and phylogenetic characterization of the ZIP family of metal ion transport proteins. J Membr Biol 166:1–7
Eren E, Argüello JM (2004) Arabidopsis HMA2, a divalent heavy metal-transporting PIB-type ATPase, is involved in cytoplasmic Zn2+ homeostasis. Plant Physiol 136:3712–3723
Fan B, Rosen BP (2002) Biochemical characterization of CopA, the Escherichia coli Cu(I)- translocating P-type ATPase. J Biol Chem 277:46987–46992
Fatemi N, Sarkar B (2002) Structural and functional insights of Wilson disease copper-transporting ATPase. J Bioenerg Biomembr 34:339–349
Forbes JR, Hsi G, Cox DW (1999) Role of the copper-binding domain in the copper transport function of ATP7B, the P-type ATPase defective in Wilson disease. J Biol Chem 274:12408–12413
Gitschier J, Moffat B, Reilly D et al (1998) Solution structure of the fourth metal-binding domain from the Menkes copper-transporting ATPase. Nat Struct Biol 5:47–54
Hamza I, Schaefer M, Klomp LW et al (1999) Interaction of the copper chaperone HAH1 with the Wilson disease protein is essential for copper homeostasis. Proc Natl Acad Sci USA 96:13363–13368
Haupt M, Bramkamp M, Coles M et al (2004) Inter-domain montions of the N-domain of the KdpFABC commplex, a P-type ATPase, are not driven by ATP-induced conformational changes. J Mol Biol 342:1547–1558
Hou ZJ, Narindrasorasak S, Bhushan B et al (2001) Functional analysis of chimeric proteins of the Wilson Cu(I)-ATPase (ATP7B) and ZntA, a Pb(II)/Zn(II)/Cd(II)-ATPase from Escherichia coli. J Biol Chem 276:40858–40863
Huffman DL, O’Halloran TV (2000) Energetics of copper trafficking between the Atx1 metallochaperone and the intracellular copper transporter, Ccc2. J Biol Chem 275:18611–18614
Hung IH, Suzuki M, Yamaguchi Y et al (1997) Biochemical characterization of the Wilson disease protein and functional expression in the yeast Saccharomyces cerevisiae. J Biol Chem 272:21461–21466
Hussain D, Haydon MJ, Wang Y et al (2004) P-type ATPase heavy metal transporters with roles in essential zinc homeostasis in Arabidopsis. Plant Cell 16:1327–1339
Jensen PY, Bonander N, Máller LB et al (1999) Cooperative binding of copper(I) to the metal binding domains in Menkes disease protein. Biochim Biophys Acta 1434:103–113
Kahn D, David M, Domergue O et al (1989) Rhizobium meliloti fixGHI sequence predicts involvement of a specific cation pump in symbiotic nitrogen fixation. J Bacteriol 171:929–939
Kaplan JH (2002) Biochemistry of Na,K-ATPase. Annu Rev Biochem 71:511–535
Larin D, Mekios C, Das K et al (1999) Characterization of the interaction between the Wilson and Menkes disease proteins and the cytoplasmic copper chaperone, HAH1p. J Biol Chem 274:28497–28504
Lingrel JB, Croyle ML, Woo AL et al (1998) Ligand binding sites of Na,K-ATPase. Acta Physiol Scand Suppl 643:69–77
Liu J, Dutta SJ, Stemmler AJ et al (2006) Metal-binding affinity of the transmembrane site in ZntA: implications for metal selectivity. Biochemistry 45:763–772
Liu J, Stemmler AJ, Fatima J et al (2005) Metal-binding characteristics of the amino-terminal domain of ZntA: binding of lead is different compared to cadmium and zinc. Biochemistry 44:5159–5167
Loudianos G, Dessi V, Lovicu M et al (1999) Mutation analysis in patients of mediterranean descent with Wilson disease: identification of 19 novel mutations. J Med Genet 36:833–836
Lowe J, Vieyra A, Catty P et al (2004) A mutational study in the transmembrane domain of Ccc2p, the yeast Cu(I)-ATPase, shows different roles for each Cys-Pro-Cys cysteine. J Biol Chem 279:25986–25994
Lutsenko S, Kaplan JH (1995) Organization of P-type ATPases: Significance of structural diversity. Biochemistry 34:15607–15613
Lutsenko S, Petrukhin K, Cooper MJ et al (1997) N-terminal domains of human copper-transporting adenosine triphosphatases (the Wilson’s and Menkes disease proteins) bind copper selectively in vivo and in vitro with stoichiometry of one copper per metal-binding repeat. J Biol Chem 272:18939–18944
Lutsenko S, Tsivkovskii R, Walker JM (2003) Functional properties of the human copper-transporting ATPase ATP7B (the Wilson’s disease protein) and regulation by metallochaperone Atox1. Ann N Y Acad Sci 986:204–211
MacLennan DH, Rice WJ, Green NM (1997) The mechanism of Ca2+ transport by sarco(endo)plasmic reticulum Ca2+-ATPases. J Biol Chem 272:28815–28818
Mana-Capelli S, Mandal AK, Argüello JM (2003) Archaeoglobus fulgidus CopB is a thermophilic Cu2+-ATPase. Functional role of its Histidine-rich N-terminal metal binding domain. J Biol Chem 278:40534–40541
Mandal AK, Argüello JM (2003) Functional roles of metal binding domains of the Archaeoglobus fulgidus Cu+ ATPase CopA. Biochemistry 42:11040–11047
Mandal AK, Cheung WD, Argüello JM (2002) Characterization of a thermophilic P-type Ag+/Cu+-ATPase from the extremophile Archaeoglobus fulgidus. J Biol Chem 277:7201–7208
Mandal AK, Yang Y, Kertesz TM et al (2004) Identification of the transmembrane metal binding site in Cu+-transporting PIB-type ATPases. J Biol Chem 279:54802–54807
Martell E, Smith R (2004) NIST Critical Stability Constants of Metal Complexes. National Institute of Standards and Technology (NIST) Standard Reference Database 46
Melchers K, Weitzenegger T, Buhmann A et al (1996) Cloning and membrane topology of a P type ATPase from Helicobacter pylori. J Biol Chem 271:446–457
Mitra B, Sharma R (2001) The cysteine-rich amino-terminal domain of ZntA, a Pb(II)/Zn(II)/Cd(II)-translocating ATPase from Escherichia coli, is not essential for its function. Biochemistry 40:7694–7699
Morgan CT, Tsivkovskii R, Kosinsky YA et al (2004) The distinct functional properties of the nucleotide-binding domain of ATP7B, the human copper-transporting ATPase: analysis of the Wilson disease mutations E1064A, H1069Q, R1151H, and C1104F. J Biol Chem 279:36363–36371
Nucifora G, Chu L, Misra TK et al (1989) Cadmium resistance from Staphylococcus aureus plasmid pI258 cadA gene results from a cadmium-efflux ATPase. Proc Natl Acad Sci USA 86:3544–3548
Odermatt A, Suter H, Krapf R et al (1993) Primary structure of two P-type ATPases involved in copper homeostasis in Enterococcus hirae. J Biol Chem 268:12775–12779
Okkeri J, Haltia T (1999) Expression and mutagenesis of ZntA, a zinc-transporting P-type ATPase from Escherichia coli. Biochemistry 38:14109–14116
Okkeri J, Laakkonen L, Haltia T (2004) The nucleotide-binding domain of the Zn2+-transporting P-type ATPase from Escherichia coli carries a glycine motif that may be involved in binding of ATP. Biochem J 377:95–105
Olesen C, Sørensen TL, Nielsen RC et al (2004) Dephosphorylation of the calcium pump coupled to counterion occlusion. Science 306:2251–2255
Outten CE, O’Halloran TV (2001) Femtomolar sensitivity of metalloregulatory proteins controlling zinc homeostasis. Science 292:2488–2492
Paulsen IT, Saier MH Jr (1997) A novel family of ubiquitous heavy metal ion transport proteins. J Membr Biol 156:99–103
Petris MJ, Mercer JF, Culvenor JG et al (1996) Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking. Embo J 15:6084–6095
Phung LT, Ajlani G, Haselkorn R (1994) P-type ATPase from the cyanobacterium Synechococcus 7942 related to the human Menkes and Wilson disease gene products. Proc Natl Acad Sci USA 91:9651–9654
Rensing C, Fan B, Sharma R et al (2000) CopA: An Escherichia coli Cu(I)-translocating P-type ATPase. Proc Natl Acad Sci USA 97:652–656
Rensing C, Ghosh M, Rosen BP (1999) Families of soft-metal-ion-transporting ATPases. J Bacteriol 181:5891–5897
Rensing C, Mitra B, Rosen BP (1997) The zntA gene of Escherichia coli encodes a Zn(II)-translocating P-type ATPase. Proc Natl Acad Sci USA 94:14326–14331
Rosenzweig AC, Huffman DL, Hou MY et al (1999) Crystal structure of the Atx1 metallochaperone protein at 1.02 A resolution. Structure 7:605–617
Rutherford JC, Cavet JS, Robinson NJ (1999) Cobalt-dependent transcriptional switching by a dual-effector MerR-like protein regulates a cobalt-exporting variant CPx-type ATPase. J Biol Chem 274:25827–25832
Sazinsky MH, Argüello JM, Rosenzweig AC (2006a) Structure of the actuator domain from the Archaeoglobus fulgidus Cu+-ATPase. Biochemistry 45:9949–9955
Sazinsky MH, Mandal AK, Argüello JM et al (2006b) Structure of the ATP Binding Domain from the Archaeoglobus fulgidus Cu+-ATPase. J Biol Chem 281:11161–11166
Schaefer M, Hopkins RG, Failla ML, et al (1999) Hepatocyte-specific localization and copper-dependent trafficking of the Wilson’s disease protein in the liver. Am J Physiol 276:G639–G646
Seigneurin-Berny D, Gravot A, Auroy P et al (2006) HMA1, a new Cu-ATPase of the chloroplast envelope, is essential for growth under adverse light conditions. J Biol Chem 281:2882–2892
Sharma R, Rensing C, Rosen BP et al (2000) The ATP hydrolytic activity of purified ZntA, a Pb(II)/Cd(II)/Zn(II)-translocating ATPase from Escherichia coli. J Biol Chem 275:3873–3878
Solioz M, Vulpe C (1996) CPx-type ATPases: a class of P-type ATPases that pump heavy metals. Trends Biochem Sci 21:237–241
Sørensen TL, Møller JV, Nissen P (2004) Phosphoryl transfer and calcium ion occlusion in the calcium pump. Science 304:1672–1675
Tottey S, Rich PR, Rondet SA et al (2001) Two Menkes-type atpases supply copper for photosynthesis in Synechocystis PCC 6803. J Biol Chem 276:19999–20004
Toyoshima C, Inesi G (2004) Structural basis of ion pumping by Ca2+-ATPase of the sarcoplasmic reticulum. Annu Rev Biochem 73:269–292
Toyoshima C, Nakasako M, Nomura H et al (2000) Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å. Nature 405:647–655
Toyoshima C, Nomura H (2002) Structural changes in the calcium pump accompanying the dissociation of calcium. Nature 418:605–611
Toyoshima C, Nomura H, Tsuda T (2004) Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues. Nature 432:361–368
Tsai KJ, Lin YF, Wong MD et al (2002) Membrane topology of the p1258 CadA Cd(II)/Pb(II)/Zn(II)-translocating P-type ATPase. J Bioenerg Biomembr 34:147–156
Tsivkovskii R, Efremov RG, Lutsenko S (2003) The role of the invariant His-1069 in folding and function of the Wilson’s disease protein, the human copper-transporting ATPase ATP7B. J Biol Chem 278:13302–13308
Tsivkovskii R, Eisses JF, Kaplan JH et al (2002) Functional properties of the copper-transporting ATPase ATP7B (the Wilson’s disease protein) expressed in insect cells. J Biol Chem 277:976–983
Tsivkovskii R, MacArthur BC, Lutsenko S (2001) The Lys1010-Lys1325 fragment of the Wilson’s disease protein binds nucleotides and interacts with the N-terminal domain of this protein in a copper-dependent manner. J Biol Chem 276:2234–2242
Verret F, Gravot A, Auroy P et al (2004) Overexpression of AtHMA4 enhances root-to-shoot translocation of zinc and cadmium and plant metal tolerance. FEBS Lett 576:306–312
Voskoboinik I, Brooks H, Smith S et al (1998) ATP-dependent copper transport by the Menkes protein in membrane vesicles isolated from cultured Chinese hamster ovary cells. FEBS Lett 435:178–182
Voskoboinik I, Mar J, Strausak D et al (2001) The regulation of catalytic activity of the menkes copper-translocating P-type ATPase. Role of high affinity copper-binding sites. J Biol Chem 276:28620–28627
Voskoboinik I, Strausak D, Greenough M et al (1999) Functional analysis of the N-terminal CXXC metal-binding motifs in the human Menkes copper-transporting P-type ATPase expressed in cultured mammalian cells. J Biol Chem 274:22008–22012
Vulpe C, Levinson B, Whitney S et al (1993) Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat Genet 3:7–13
Walker JM, Huster D, Ralle M et al (2004) The N-terminal metal-binding site 2 of the Wilson’s Disease Protein plays a key role in the transfer of copper from Atox1. J Biol Chem 279:15376–15384
Walker JM, Tsivkovskii R, Lutsenko S (2002) Metallochaperone Atox1 transfers copper to the NH2-terminal domain of the Wilson’s disease protein and regulates its catalytic activity. J Biol Chem 277:27953–27959
Wernimont AK, Huffman DL, Lamb AL et al (2000) Structural basis for copper transfer by the metallochaperone for the Menkes/Wilson disease proteins. Nature Struct Biol 7:766–771
Williams LE, Mills RF (2005) P1B-ATPases–an ancient family of transition metal pumps with diverse functions in plants. Trends Plant Sci 10:491–502
Wimmer R, Herrmann T, Solioz M et al (1999) NMR structure and metal interactions of the CopZ copper chaperone. J Biol Chem 274:22597–22603
Woeste KE, Kieber JJ (2000) A strong loss-of-function mutation in RAN1 results in constitutive activation of the ethylene response pathway as well as a rosette-lethal phenotype. Plant Cell 12:443–455
Yoshimizu T, Omote H, Wakabayashi T et al (1998) Essential Cys-Pro-Cys motif of Caenorhabditis elegans copper transport ATPase. Biosci Biotechnol Biochem 62:1258–1260
Acknowledgements
We thank Matt Sazinsky, Danielle DeOssie, and Amy Rosenzweig for critical reading of this manuscript. This work was supported by NSF grant MCM-0235165 (J. M. A.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Argüello, J.M., Eren, E. & González-Guerrero, M. The structure and function of heavy metal transport P1B-ATPases. Biometals 20, 233–248 (2007). https://doi.org/10.1007/s10534-006-9055-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-006-9055-6