
Abstract
Mechanisms of soil organic carbon (C) and nitrogen (N) stabilization are of great interest, due to the potential for increased CO2 release from soil organic matter (SOM) to the atmosphere as a result of global warming, and because of the critical role of soil organic N in controlling plant productivity. Soil proteins are recognized increasingly as playing major roles in stabilization and destabilization of soil organic C and N. Two categories of proteins are proposed: detrital proteins that are released upon cell death and functional proteins that are actively released into the soil to fulfill specific functions. The latter include microbial surface-active proteins (e.g., hydrophobins, chaplins, SC15, glomalin), many of which have structures that promote their persistence in the soil, and extracellular enzymes, responsible for many decomposition and nutrient cycling transformations. Here we review information on the nature of soil proteins, particularly those of microbial origin, and on the factors that control protein persistence and turnover in the soil. We discuss first the intrinsic properties of the protein molecule that affect its stability, next possible extrinsic stabilizing influences that arise as the proteins interact with other soil constituents, and lastly controls on accessibility of proteins at coarser spatial scales involving microbial cells, clay particles, and soil aggregates. We conclude that research at the interface between soil science and microbial physiology will yield rapid advances in our understanding of soil proteins. We suggest as research priorities determining the relative abundance and turnover time (age) of microbial versus plant proteins and of functional microbial proteins, including surface-active compounds.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Mechanisms of soil organic carbon (C) and nitrogen (N) stabilization are currently of great interest, due to the potential for increased CO2 release from soil organic matter (SOM) to the atmosphere as a result of global warming, and the critical role of soil organic N in controlling plant productivity. The global pool of soil organic N is about 1017 g versus 1023for the lithosphere and 1021 for the atmosphere (Paul and Clark 1996). The mean residence time of N in the soil has been calculated as 50 years versus 26 years for C (Schlesinger 1991). Although part of this difference is almost certainly due to reprocessing of N by soil microorganisms, the mechanisms that stabilize N-containing organic compounds may also differ in part from those that stabilize non-N-containing organic compounds. More recently, direct measurements of chirality of several amino acids have suggested ages for soil protein in the range of hundreds of years (Amelung et al. 2006).
Organic compounds may persist in soil as a result of their inherent chemical recalcitrance, inaccessibility due to physical protection, or stabilization due to intermolecular interactions with minerals, inorganic solutes, and other organic compounds (e.g., Christensen 1992; Sollins et al. 1996). However, the relative importance of these mechanisms for stabilization of nitrogenous organics in soils is not well explored. The majority of the identifiable soil organic N occurs as amino (or more precisely amide) compounds (see Table 1), based on both direct extraction from soils (Bremner 1965; Leinweber and Schulten 2000; Rillig 2004) and 15N-NMR studies (Knicker 2000; Smernik and Baldock 2005). The two main categories of amino-N compounds are the intact proteins released for various extracellular functions (surface-active agents, extracellular enzymes) and detrital proteins and polypeptides—plant and microbial (and some animal) constituents in various stages of transformation. Also present in the soil are amino sugars and compounds formed by abiotic interactions, such as protein–tannin complexes and Maillard reaction products, as well as various heterocyclic pyrolysis products (see Knicker 2006, this volume).
Figure 1 presents a simplified conceptual model of the soil amino-N cycle. Biota, mainly plants and microbes, release diverse protein, peptide, and amino N substrates upon cell death and by active exudation. Once outside the protective cell, proteins and peptides are susceptible to breakdown via processes that can include hydrolysis by extracellular microbial enzymes or ingestion by soil fauna. Subsequent fates include leaching and gaseous loss as well as uptake by plants and microbes. One particularly important fate of decomposed protein (inorganic N and amino acids) is subsequent uptake and re-synthesis into microbial protein (Miltner and Zech 1999).

Depiction of a simplified soil protein cycle, relating sources and fates of proteins. Proteins can be either decomposed, or they can be stabilized in soils (see Fig. 2), if only transiently. In their stabilized form they can either be inactive or active with respect to their original cellular/organismic function. Stabilized proteins are repeatedly re-exposed to the environmental filter once they become accessible to enzymes
A significant amount of protein, however, enters the soil matrix relatively unaltered and is stabilized for some time against microbial degradation. Many of these now-extracellular proteins no longer express their original function (e.g., photosynthetic enzymes), but some can be completely functional. The latter potentially include surface-active proteins such as hydrophobins, as well as those extracellular enzymes active in decomposition. Moreover, it seems reasonable to expect that proteins that can retain their functionality may be especially resistant to degradation (recalcitrant).
In the following sections we attempt to relate persistence of proteins in soil first to their intrinsic molecular properties, then to extrinsic intermolecular interactions between proteins and other soil constituents, and lastly to their occurrence within microbial cells and soil aggregates. We then introduce several groups of specialized microbial proteins that may perform specific functions in the soil matrix and have seen recent intensive scrutiny in microbial physiology/biochemistry. We present hypotheses about the role of these specialized functional proteins in soil and finish with suggestions for future research priorities and opportunities.
Intrinsic stabilization of proteins
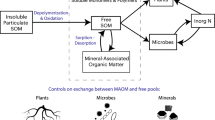
Several characteristics and processes may increase protein resistance to degradation by altering their structure to occlude the peptide bond (Fig. 2).

Conceptual overview of structures and scales involved in microbial protein stabilization in soils. The various processes occurring at different scales in this hierarchical framework are discussed in detail in the text. The listed size ranges may overlap between different structures and are only intended to serve as general guides
For example, amyloid aggregates and resultant fibrils are two related features of protein structure, known largely from medicine where misfolded proteins are responsible for at least 21 major diseases (e.g., Alzheimer’s; Merlini and Belloti 2003). Proteins are inherently subject to some degree of misfolding, resulting in the formation of amyloid aggregates that can form intertwining rope-like fibrils (Dobson 1999; Gebbink et al. 2005). The extent of amyloid aggregate and fibril formation can be affected by extraneous factors, including temperature, ligands, and the nature of specific peptides. Details of the molecular mechanisms by which proteins misfold and aggregate to form intertwining fibrils are given by Alexandrescu (2005), Ho et al. (2005), and Merlini and Belloti (2003). Amyloid fibrils and aggregates are very effectively stabilized against chemical denaturation and enzymatic hydrolysis. Although the occurrence of such fibrils and aggregates in soil is virtually unstudied, several of the soil microbial proteins discussed in detail below are known to exist mostly as amyloid fibrils (e.g. hydrophobins).
Glycosylation, the covalent linkage of specific oligosaccharides to specific amino acids, is an enzymatically mediated intracellular modification of proteins that occurs prior to secretion (Varki 1993). Glycosylation is known to increase in vitro protein stability against proteolytic enzymes up to ten-fold (Bernard et al. 1983; Opdenakker et al. 1993; Varki 1993; West 1986). The extent to which soil proteins are stabilized by glycosylation remains unknown but merits attention.
Extrinsic stabilization of proteins: interactions with other soil organic compounds
The preceding section focused on intrinsic molecular properties of proteins that might contribute to their persistence in the soil. Protein stability can be further enhanced by interactions with other soil molecules, specifically polyphenolics and carbohydrates.
Polyphenolics
Probably the oldest known mechanism of protein stabilization involves those plant polyphenols commonly referred to as tannins. Tannin structures, reactivity, and possible roles in decomposition and nutrient cycling have been extensively reviewed (Zucker 1983; Horner et al. 1988; Hättenschwiler and Vitousek 2000; Kraus et al. 2003a, b; Nierop et al. 2006). Proteins can react with tannins and related polyphenols to form soluble and insoluble products through reversible non-covalent processes such as hydrogen bonding and hydrophobic interactions (Loomis and Battaile 1966; Oh et al. 1980; McManus et al. 1981; Nyman 1985; Hagerman et al. 1998). The amount and solubility of these complexes and their resistance to enzyme hydrolysis vary extensively with type of protein and tannin, ratio of protein to tannin, ionic strength and pH. (Basaraba and Starkey 1966; Benoit et al. 1968; Lewis and Starkey 1968; Hagerman and Robbins 1987). Covalent bonds can be formed by nucleophilic addition between quinones and N or S nucleophiles (Loll and Bollag 1983; Haslam 1989).
Virtually all of our understanding of tannin–protein interactions comes from laboratory experiments. In vitro, tannins are known to slow protein degradation in artificial systems using soil or soil inoculum. Basaraba and Starkey (1966) reported decomposition of tannin–gelatin mixtures was inhibited 20%, 69% and 52% by tannin:protein mixtures of 1:4, 1:1 and 2:1, respectively. Inhibition of ammonification of gelatin-litter extract precipitates by a soil inoculum ranged from 14% to 85% across litter from 14 tree species (Howard and Howard 1993). Inhibition of protein decomposition varied from 18% to 70% for combinations of two tannins and four proteins (Lewis and Starkey 1968). Field-based evidence that tannins stabilize soil proteins remains largely circumstantial at this time, but a stabilizing role is implied by relations between litter chemistry and rates of N mineralization (Northup et al. 1995, 1998; Fierer et al. 2001; Kraus et al. 2003a and references therein).
In contrast with numerous reports of tannins in green leaves and litter (see Kraus et al. 2003a, b), there are relatively few values for tannins in soil horizons (Table 2). While these data are of little overall comparative value because of differences in extraction and analysis methods, a few generalizations are possible. Amounts of condensed tannins in organic horizons vary with plant species (Kuiters and Denneman 1987; Northup et al. 1995; Smolander et al. 2005), stand age (Lorenz et al. 2000), and successional stage (Gallet and Lebreton 1995; Northup et al. 1995). Finding tannins in the more decomposed organic horizons is not always successful: Fierer et al. (2001) found no condensed tannins (CT) in the Oe horizons of Populus or Alnus stands that contained 106.4 and 0.9 mg CT g−1 , respectively, in the Oi layers. The only successful extractions of tannic acid from mineral soils have shown complex patterns with depth, vegetation and successional stage (Blum and Rice 1969; Rice and Pancholy 1973).
Experimental addition of tannins to soil has yielded little to no recovery. Bradley et al. (2000) recovered 0.32% and 1.16% respectively of Abies and Kalmia tannins added to a black spruce organic horizon (3% wt/wt) and concluded that much of “the tannins may have become tightly bound to organic matter, including protein”. Schofield et al. (1998) tried several extraction and detection methods but were unable to detect Salix-derived condensed tannins that had been added to mineral soil; they suggested that attachment to soil particles had made the tannins unrecoverable. Despite abundant evidence for protein in soil (Table 1), and limited evidence for tannins in soil organic and mineral horizons (Table 2), we found no direct evidence for the existence of protein-tannin complexes in soil.
Another common plant phenolic that can interact with protein is lignin. Waksman and Iver (1932) found that alkali lignin could remove up to 33% of casein-N from solution. Using a mixture of soil microorganisms, ammonification of casein was reduced 25% in 11 days by mechanical mixing with lignin and by over 95% after dissolution and reprecipitation with lignin. This led to a model of SOM in which a major portion consists of ligno-protein condensation products (Waksman 1938). This model was later replaced by a more encompassing “polyphenol” model (Stevenson 1994) in which quinones can covalently bond to protein N. This model is now being replaced in turn by the concept of the supra-molecular assemblage dominated by amphipathics (Piccolo 2001; Sutton and Sposito 2005; Kleber et al. 2006, this volume). Formation of covalent and other bonds between proteins and lignin residues could occur by the same mechanisms suggested above for tannin–protein interactions. The importance of these mechanisms may have been largely overlooked because lignin becomes less distinguishable and more soluble with progressive decomposition.
Fungal melanins are polyphenolic pigments that are synthesized by certain fungi either as constituents of the cell wall or as exudates (Coelho et al. 1985; Butler and Day 1998) and that can then interact act with proteins in much the same way as plant tannins. Kuo and Alexander (1967) found that protease hydrolysis of a casein-melanin mixture over two hours was 44.3% of casein alone. Although melanized hyphae, as well as fungal resting structures (sclerotia), are found universally in soils, their mass and persistence remain unknown. Possible stabilization within fungal biomass is discussed below.
Carbohydrates
In addition to the intrinsic protein stabilization through glycosylation, proteins may also be stabilized through extrinsic interactions with soil carbohydrates. Glycation, or the Maillard reaction, is the non-enzymatic covalent bonding of a sugar aldehyde to an amino group, especially the side-chain amino groups of lysine and arginine (Ikan 1996). Glycation has been shown to significantly increase the stability of proteins and peptides in laboratory studies. Gil et al. (1991) reported that glycation could inhibit enzymatic breakdown of proteins. Jakas and Horvat (2004) found enzymatic decomposition half-life of a glycated pentapeptide to be over 50 times greater than that of the non-glycated form. While the Maillard reaction is well known in medicine and nutrition (Ikan 1996), the extent to which it can stabilize or protect proteins in soils has not been studied. Previously considered to require too extreme a temperature to occur extensively in soils (Arfaioli et al. 1999; Bosetto et al. 2002), recent in vitro work has shown that, at least for free amino acids, the Maillard reaction can be catalyzed by clays (Arfaioli et al. 1999; Bosotto et al., 2002), common minerals (Jokic et al. 2001) and polyphenols (Jokic et al. 2004). Maillard products occur extensively in soil char, however (review by Knicker (2006, this volume).
Phytates, another group of potential protein-complexing carbohydrates, consist of a sugar core (inositol) in which each hydroxyl is phosphorylated. Unlike the strong covalent bonding found in glycation and glycosylation, phytate–protein complexes are formed through weaker electrostatic bonds between the negatively charged phosphates and positively charged basic amino acids, as well as possibly through cation-bridging of phosphates to carboxylates (Cosgrove 1966; Anderson 1985). Widely studied in nutrition, phytate-protein complexes resist proteolysis (Cheryan 1980; Ravindran et al. 1995). For example, phytates inhibited digestion of casein by trypsin by 45% (Singh and Krikorian 1982). Although phytates are the most abundant form of organic phosphorus in soils (Dalal 1977), the extent of their interaction with soil protein has not been studied.
“Humic”–protein interactions
“Humic” substances are an operationally defined soil fraction. The nature of these materials is undergoing a major redefinition from a complex poly-condensed macromolecular structure (Schulten and Schnitzer 1993) to a less strongly bonded dynamic complex of smaller distinct molecules that include mainly plant and microbial constituents and their partial decomposition products held together by H-bonding, hydrophobic interactions and covalent bonds (Burdon 2001; Piccolo 2001; Sutton and Sposito 2005; Kleber et al. 2006, this volume;). The presence of protein in humic extracts has been implied by release of amino acids by acid hydrolysis (Bremner 1965) and proteolytic enzymes (Ladd and Brisbane 1967; Jahnel and Frimmel 1995). Knicker and Hatcher (1997) and Zang et al. (2000) have suggested that proteins might be provided long-term protection by their incorporation into hydrophobic domains of soil organic matter. For example, by measuring mineralization of 14C-labeled protein, Verma et al. (1975) found that mixtures of protein and “model” humic polymers decomposed at 37% the rate of protein alone. Covalently bonding of protein with “model” polymers decreased decomposition by phenoloxidase by 88% over 12 weeks. A similar experiment in soil showed that mixing of labeled protein and model humic polymers reduced protein decomposition by up to 76% over 12 weeks (Martin et al. 1978). Recent NMR-based evidence for the in vitro covalent coupling of peptides to phenolic components of SOM (Hu and Hatcher 2003) suggests a way to find covalent phenolic-protein condensation products in nature.
Protein interactions with mineral surfaces
Peptidic compounds in general sorb strongly to a wide variety of clays (see Theng 1979), with strength of bonding varying over several orders of magnitude depending on the protein. This process is used in the wine industry to remove protein (fining) and in a variety of other commercial applications. The mechanisms of such sorption have long been thought to be primarily electrostatic in nature, but current research is suggesting that ligand exchange and physisorption may be equally important. This important mechanism of protein stabilization is reviewed extensively in a companion paper (Kleber et al., 2006, this volume).
Physical protection of proteins (changes in accessibility)
Although there is little specific research on mechanisms of physical protection of proteins in soil, we provide here a brief overview of the general mechanisms that operate on all SOM and thus can be expected to promote protein stabilization.
Microbial biomass and biofilms
The presence of proteins within the cytoplasm of microbial biomass inherently imparts some degree of protection which can vary with the nature of the microorganism. Nakas and Klein (1979) found that bacterial cells decomposed faster than fungal hyphae in a grassland soil. Among the fungi, non-pigmented (hyaline) cells decompose faster than dark-pigmented (melanized) cells (Kuo and Alexander 1967; Lockwood 1960). Proteins are also significant internal components of fungal hyphal walls (see section Hydrophobins), where, in melanized fungi, they can be further protected from proteolytic enzymes (Butler and Day 1998). While these proteins may not be afforded long-term protection, owing to the turnover of microbial biomass, they are nevertheless not immediately released into the soil solution. Instead, upon cell death they may be deposited into soil micropores as covalently linked components within the hyphal (or bacterial) wall (Driver et al. 2005).
Biofilms, a topic for research across many fields (Parsek and Fuqua 2004), have been observed in soils (Grossman and Lynn 1967; Harris 1972). However, only recently are we appreciating their ecological roles, for example in terms of formation of consortia of bacteria associating with mycorrhizal fungal hyphae and roots (Sen 2003). Biofilms contain not only living cells (representing mixed populations of microbes) but also a non-living matrix consisting of what has been termed extracellular polymeric substances, which can also contain significant protein (Omoike and Chorover 2004; Sternberger and Holden 2004). Soil biofilms also represent a special case of sorption to mineral surfaces in that the first stage in biofilm formation involves attachment of specialized extracellular proteins (Bashan and Levanony 1988). The extent to which occlusion within biofilms can protect and stabilize protein N in soils is unknown, although circumstantial evidence comes from the requirements for enzymatic, chemical and mechanical treatments to disrupt biofilms (Brisou 1995; Böckelmann et al. 2003).
Soil aggregation and soil physical structure
Proteins, like any other organic compounds, can only be degraded if they are accessible to microbes or extracellular enzymes (Fig. 2). Accessibility, in turn, depends on physical location within the soil fabric, and all of the above-mentioned processes occur within the context of this framework. The strength of this fabric is not uniform, and when the soil is subject to any physical disruption it fractures along planes of weakness (Díaz-Zorita et al. 2002). These planes define aggregates, assemblages of mineral particles, organic debris, and amorphous materials that can vary in size from submicron to several centimeters (Fig. 2). Permanent binding agents are responsible for the stabilization of small microaggregates (<30 μm), whereas macroaggregates (>250 μm) are bound predominantly by transient binding agents originating from roots and hyphae (Tisdall and Oades 1982; Oades 1984; Six et al. 2004). Aggregates exist in what can be conceptualized as a hierarchical order, with microaggregates (<250 μm) bound and often formed within macroaggregates (Oades 1984; Angers et al. 1997). Microaggregates turn over much more slowly than macroaggregates and thus provide longer-term stabilization (Six et al. 1998). Six et al. (2002) compared organic C and N mineralization from crushed versus intact aggregates and concluded that, in both tropical and temperate soils, organic N (which would include an unknown proportion of protein) and C was more strongly protected within microaggregates.
Access to substrates requires movement of organisms through pores or of enzymes through water films (Elliott and Coleman 1988; Chenu and Stotzky 2002). Pores exist even in the smallest microaggregates (indeed, even in otherwise solid mineral particles) and even a single small pore neck can greatly impede access through an otherwise quite large and continuous pore. Additionally, physiochemical characteristics such as oxygen concentration can differ drastically between the interior and exterior of aggregates (Sexstone et al. 1985). Such gradients provide an additional mechanism for stabilization by dictating the nature and size of the microbial populations at each location (Mummey et al. 2006; Blackwood et al. 2006) and what metabolic processes are possible.
Aggregates are dynamic. They form and reform over time thereby making the organic material occluded within them accessible to degradative enzymes (Six et al. 1998; Plante and McGill 2002; Plante et al. 2002; DeGryze et al. 2005). Tillage increases this turnover rate, which is generally faster for small aggregates than for large (Six et al. 1998, 2004).
Proteins, like any other organic substrate in soil, can occur anywhere in the soil fabric, thus may be subject to any degree of accessibility. Brewer (1964) published the first comprehensive system for describing soil fabric. Although his work has given rise to a large body of literature on soil fabric in relation to mineralogy and management practices, we know of no papers relating this to organic N turnover in soils. Likewise, there is an extensive and rapidly growing literature on the nature of and controls on soil aggregate formation (e.g., Jastrow 1996, Six et al. 2002, 2004, Rillig and Mummey 2006), and on the effects of aggregation and aggregate stability on C turnover (Six et al. 1998). The literature on effects on organic N turnover is sparser and we know of no papers directly relating aggregation specifically to protein persistence.
Specific microbial proteins
Having discussed general mechanisms of protein stabilization, we next review relevant intrinsic properties of specific microbial proteins, and present hypotheses as to their abundance and persistence in soils. In our discussion we include hydrophobins, SC15, repellents, and glomalin(s), which are examples of fungal proteins, as well as chaplins, examples of bacterial structural proteins.
Hydrophobins
Hydrophobins are secreted by ascomycetes and the basidiomycetes (Wösten 2001). Many fungi belonging to these two phyla of the eumycotan kingdom contain multiple hydrophobin genes, and the encoded proteins fulfill a wide spectrum of functions (Wösten 2001). For instance, they enable fungi to escape the aqueous environment to grow into the air, confer hydrophobicity to fungal aerial structures such as fruiting bodies and spores, and mediate attachment of fungi to hydrophobic solids.
Hydrophobins are about 100 amino acids in length (Wösten 2001) and can make up ten percent of the total cellular protein (Wessels et al. 1991a, b). They are not highly similar but share eight conserved cysteine residues. Based on solubility characteristics and hydropathy patterns, Wessels (1994) discriminated between class I and class II hydrophobins. The latter hydrophobins, which are only produced by the ascomycetes, may have evolved independently from the class I hydrophobins (Whiteford and Spanu 2002). Both class I and class II hydrophobins organize themselves into an amphipathic two-dimensional protein film at hydrophobic–hydrophilic interfaces such as those between water and air, water and oil, or water and a hydrophobic solid like Teflon (Wösten and de Vocht 2000). The protein film of the class II hydrophobins has been suggested to be composed of packed tetramers, at least in case of HFBII of Trichoderma reesei (Torkkeli et al. 2002). The film is not very stable. It dissociates upon applying pressure or adding ethanol or diluted detergent (Carpenter et al. 1992; Russo et al. 1982; Takai and Richards 1978; SA Askolin & HAB Wösten, unpublished). Similarly, class II hydrophobins detach from a hydrophobic solid upon treatment with hot detergent or even washing with water (Linder et al. 2002; SA Askolin & HAB Wösten, unpublished).
In contrast to the films of class II hydrophobins, those of class I are highly insoluble. They only dissociate into the water-soluble form upon treatment with formic acid (Wessels et al. 1991a, b) or trifluoroacetic acid (de Vries et al. 1993). This class I hydrophobin film consists of a mosaic of aligned 10 nm wide fibrils. These hydrophobin fibrils are called rodlets (Wösten et al. 1993) and have an amyloid-like nature (Wösten and de Vocht 2000; Butko et al. 2001). The hydrophobin rodlets interact and form a physically strong membrane that can span a gap of a few millimeters in diameter (de Vocht et al. 2002) or prevent a water droplet from being sucked into a Pasteur pipette (Lugones et al. 2004).
Membranes formed by class I hydrophobins have remarkable properties. They are not only highly insoluble (Wessels et al. 1991a, b), semi permeable (Wang et al. 2005), and protease resistant (MI Janssen and HAB Wösten, unpublished results), they are also among the most surface-active aggregates in nature. In fact, with a maximal lowering of the water surface tension from 72 to 24 mJ m−2, the SC3 hydrophobin is the most surface active protein known (Wösten et al. 1999). Moreover, the amphipathic membranes of class I hydrophobins can turn hydrophilic surfaces hydrophobic and vice versa (Wösten et al. 1993, 1994, 1995; Lugones et al. 1996, 1998, 1999). These changes in the physico-chemical properties are stable since assembled class I hydrophobin strongly adheres to hydrophobic solids and to some hydrophilic surfaces as well (Wösten and de Vocht 2000). The hydrophobic side of class I hydrophobin membranes (exposed after assembly at a hydrophilic surface) is invariable strongly water repellent. With a water contact angle of about 110° it is as hydrophobic as Teflon. In contrast, the hydrophilic side of class I hydrophobin membranes (exposed after assembly at a hydrophobic solid) is variable in wettability. Water contact angles range between 36° and 63° depending on the hydrophobin used.
We do not yet have any information on the occurrence or behavior of hydrophobins in soil. However, class I hydrophobins are more likely to contribute to soil C and N stabilization than class II hydrophobins, since the former assemble in a highly stable film. Class I hydrophobins are secreted into the aqueous environment and assemble at the hyphal surface when exposed to air or a hydrophobic solid. In the latter case, hydrophobins attach the hypha to the solid, thus potentially contributing to stabilization of aggregates. Hydrophobins secreted into the moist environment could also assemble at hydrophobic soil particles. These surfaces would become hydrophilic, thus changing the physico-chemical properties of the particles. As a result, adherence of soil bacteria and fungi may be stimulated or decreased. For example, growth and adherence of fibroblasts to Teflon could be improved by coating a hydrophobic solid with hydrophobins (Scholtmeijer et al. 2002; Janssen et al. 2002, 2004). Enzymes secreted by microorganisms may also be stabilized by the hydrophobin film. It was recently shown that loss of enzyme activity could be prevented by adsorbing enzymes to a hydrophobin-coated hydrophobic solid instead of a bare hydrophobic surface (Corvis et al. 2005). The hydrophobin coating probably prevents denaturation at the surface of the solid.
Apart from coating hydrophobic particles, hydrophobins could also assemble at hydrophilic particle surfaces, thus making them hydrophobic. Assembly at hydrophilic surfaces may occur when soils dry out and an interface is created between the soil particles and the air. The extremely hydrophobic nature of the exposed side of the hydrophobin membrane may stabilize air channels in soil aggregates by preventing capillary transport of water.
Can hydrophobins contribute significantly to soil surface area? The wood-rotting fungus Schizophyllum commune secretes up to 60 mg of SC3 hydrophobin per liter of minimal medium. This amount would be sufficient to coat 40 m2of surface (Wösten et al. 1994). Given the very large surface area of soil, it remains to be seen if the resident soil fungal biomass can produce enough of this compound to coat a significant percentage of surfaces in this environment. It also remains to be established whether this amount of hydrophobin is even secreted under natural conditions, but we do know that SC3 is produced (de Jong 2006). Since S. commune has at least four hydrophobin genes (Wessels et al. 1995), other hydrophobins may be produced under these conditions as well.
Many of these properties suggest roles in C, N and aggregate stabilization especially at the microaggregate scale. Additionally, however, since hydrophobins will likely be acting in concert with fungal hyphae, they would also be expected to make direct contributions to macroaggregates.
SC15 and repellents
As mentioned, hydrophobins have so far only been identified in ascomycetes and basidiomycetes. There is no evidence that they are produced by Glomeromycota (arbuscular mycorrhizal fungi, AMF) or Zygomycota. Possibly, these fungi have evolved other proteins with properties similar to those of hydrophobins (see below). Such proteins have been identified in basidiomycetes. The 17 kDa SC15 protein of the basidiomycete S. commune can partly substitute for the SC3 hydrophobin by reducing the water surface tension and by making aerial hyphae hydrophobic (Lugones et al. 2004). But there are no indications that SC15, which has a hydrophilic N-terminal half and a hydrophobic C-terminal half, self-assembles in a protein film and that it has affinity for hydrophobic solids. The same holds for the repellents of the heterobasidiomycete Ustilago maydis. Repellents are cell-wall located peptides of 35–53 amino acids that result from cleavage of the precursor protein Rep1 in the endoplasmic reticulum (Wösten et al. 1996). Deletion of rep1affected formation of aerial hyphae, surface hydrophobicity, and attachment to hydrophobic surfaces. In contrast, deleting either or both hydrophobin genes of U. maydis only affected aerial hyphae formation (HJ Deelstra, WR Teertstra, HAB Wösten, unpublished). From these results it is concluded that hydrophobins of U. maydis have been functionally replaced, at least partially, by repellents and possibly other molecules as well. How these proteins mediate surface hydrophobicity and attachment is not known, and it is also presently unknown if they occur in soils.
Glomalin(s)
The path of research for hydrophobins and glomalin has been exactly opposite (Rillig 2005). While there is a wealth of data available for hydrophobins from a molecular biology and biochemical perspective, there is little environmental data. Conversely, for glomalin, and glomalin-related soil proteins (Rillig 2004), the origin of research has been in soil science, and only recently has the molecular biology of the protein begun to be revealed.
Glomalin is produced by AMF, and is currently quantified from soil following an operational definition (Wright and Upadhyaya 1996) as glomalin-related soil protein (GRSP; Rillig 2004). The main detection tool is a monoclonal antibody (MAb32B11), raised originally against crushed spores of an AMF (Wright and Upadhyaya 1996). It has recently become apparent through spiking experiments that at least the Bradford-reactive soil protein fraction of GRSP likely includes proteins of non-AMF origin, contrary to previous assumptions (Rosier et al. 2006). Keeping these limitations in mind, GRSP often amounts to several (generally <5%) percent of soil C (e.g., Rillig et al. 2001), and it appears to persist in a variety of soil (years to decades; Rillig et al. 2003; Steinberg and Rillig 2003; Harner et al. 2004). Possibly partly as a consequence of its environmental persistence, and partly due to its purported role in stabilizing aggregates (Rillig and Mummey 2006), GRSP concentrations in soils are highly positively correlated with soil aggregate water stability (Wright and Upadhyaya 1998; Harner et al. 2004; Rillig 2004). As a consequence of this correlation, much research has been dedicated to defining environmental factors to which GRSP concentrations react sensitively, including management factors (reviewed in Rillig 2004) and factors of global change (e.g., Rillig et al. 1999). As opposed to the hypothesized role in soil ecology, a function of glomalin in the life history of AMF has been less clear. Driver et al. (2005) showed that glomalin was contained primarily (∼80% of the total) in the fungal mycelium, rather than secreted into the culture medium. This suggested that there is in fact a primary role for the protein in the living fungus, including a possible structural role. Indeed, recently, the putative gene for glomalin from the AMF Glomus intraradices has been isolated, sequenced and expressed, and it shows high amino acid similarity to heat shock protein 60 (Gadkar and Rillig 2006). However, it cannot yet be discounted that the Glomeromycota-produced glomalin functions also similarly to hydrophobins in other fungal groups.
In summary, unlike many of the other proteins discussed here (hydrophobins, SC15, repellents, etc.), it seems evident that glomalin is produced under sterile laboratory in vitro culture (Driver et al. 2005), as well as in the soil. Until recently research to determine how glomalin interacts with different soil constituents has been impeded by lack of the purified protein. Successful expression of the glomalin gene will make these studies possible in the near future.
Chaplins—bacterial structural protein
Like fungi, streptomycetes are abundant in soil. These Gram-positive bacteria have a life-cycle similar to that of fungi. After a feeding mycelium has been established, spore forming structures develop in the air. Chaplins of streptomycetes fulfill functions similar to those of the fungal hydrophobins. They mediate attachment of hyphae to hydrophobic surfaces (Claessen 2004), allow hyphae to escape the aqueous environment to grow into the air (Claessen et al. 2003) and make surfaces of aerial hyphae and spores hydrophobic (Claessen et al. 2003, 2004).
S. coelicolor contains eight chaplins. The mature forms of five of these chaplins (ChpD-H) are about 55 amino acids in length, while ChpA-C consist of approximately 225 amino acids (Claessen et al. 2003; Elliot et al. 2003). The latter chaplins contain two chaplin domains (i.e. sequences similar to those of mature ChpD-H) and are probably covalently linked to the cell wall via a cell wall anchoring domain. The small chaplins were found in the medium and at the surface of aerial hyphae and spores. Like hydrophobins, these chaplins self-assemble at the water-air or cell wall-air interface into a surface active rigid two-dimensional protein film that consists of amyloid-like fibrils (Claessen et al. 2003). These fibrils are also very stable and only dissolve in trifluoroacetic acid (TFA). In contrast to hydrophobins, chaplins assemble in solution when a seed of the assembled form of the protein is added. Thus, self-assembly of chaplins becomes independent of a hydrophilic–hydrophobic interface once a nucleus of amyloid has been formed.
Water-soluble chaplins do not spontaneously assemble at a hydrophobic solid (Claessen 2004). Instead, they seem to be arrested in an intermediate state of the assembly process. Heating in diluted detergent induces the protein to proceed to the amyloid form. Possibly, streptomycetes secrete molecules that induce the intermediate form of chaplins adsorbed to a hydrophobic solid to adopt the stable amyloid form.
Clearly, the properties of chaplins indicate that they could function like hydrophobins in soils. However, perhaps this would primarily occur at smaller aggregate scales (microaggregates), owing to the smaller spatial scales at which streptomycetes operate compared to fungi.
Microbial extracellular enzymes
Another group of microbial proteins that could be expected to have adapted for persistence in the soil matrix are those epi- and extracellular enzymes necessary for the processing of macromolecules into assimilable subunits. Soil enzymes have been extensively studied (Burns 1978; Burns and Dick 2002) and are without question the most researched of all functional soil proteins. Of the various sources of extracellular soil enzymes, active secretion by microbial decomposers is probably most important, as this is directly related to substrate availability and to the nutritional needs of the microbial community (Caldwell 2005). The persistence of enzymatic function once released from the cell is well-known, where the very mechanisms cited above to stabilize soil proteins, can facilitate the continuing enzymatic functions. Active humic–enzyme associations have been isolated from soils (e.g., Busto and Perez-Mateos 1995) and can be more resistant to proteolysis than the free enzyme (e.g., Sarkar and Burns 1984).
Conclusions, caveats and future research needs
Much of our discussion has centered on soil proteins in general, but it is obvious that more significant progress can be made by developing and applying tools that can discern sources and fates of specific proteins. The differential persistence of proteins of different origins and function is a source of much speculation; for example, it is often assumed that microbial proteins persist longer than plant- or animal-derived proteins. This is a testable hypothesis, and we suggest one that should be given high priority in the context of soil C and N storage. One possible approach to this is the recently emerging field of soil proteomics (Schulze 2005; Schulze et al. 2005) in which proteins recovered from environmental samples can be identified.
We have pointed out numerous gaps in our knowledge on the role of proteins in C and N stabilization. One of the most fundamental of these is our ability to quantify soil protein or peptidic N. Martens and Loeffelmann (2003) have shown that the long relied-upon HCl hydrolysis may not hydrolyze all peptidic N. Leinweber and Schulten (2000) found that peptidic N can be occluded in amorphous metal oxides and thus resist acid hydrolysis. While protein estimation by the Bradford dye reaction (Bradford 1976) is popular in various extraction schemes, the assay has a very wide response to different proteins and to different size proteins and peptides. For example, the Bradford assay responds very poorly to low molecular-weight peptides (Friedenauer and Berlet 1989), which marine research field has shown to represent the vast majority (up to 90%) of peptidic N (Sommerville and Preston 2001). Martens and Loeffelmann (2003) also suggested that the soil materials hydrolyzed with methane sulfonic acid could be of substantially smaller size than plant proteins.
While most studies have sought to determine the total amount of protein in soil by maximizing recovery (e.g., Martens and Loeffelmann 2003), it is critical to our future understanding and ultimate ability to model soil protein dynamics to distinguish soil protein fractions of differing labilities. We have reviewed a number of intrinsic and extrinsic mechanisms by which proteins can be stabilized. However, virtually all of this work is based on laboratory or microcosm studies, which are markedly affected by reactant concentrations and reaction conditions. Determining which reactions actually occur in situ and their relative contributions to soil protein persistence will be an ambitious undertaking. The notable exception to this is the growing work done with extractible glomalin-related soil protein.
Peptidic compounds in general are probably much more persistent and play a larger role in C stabilization than previously thought, due in large part to their ability to interact with a wide range of both organic and mineral surfaces (see Kleber et al. 2006, this volume). Yet none of the controls on protein persistence have been researched very extensively, and turnover rates may change with disturbance and anthropogenic influences (such as warming). One priority in this context would be better assessment of the relative importance of the various groups of mechanisms we have discussed. For example, it is possible that intrinsic properties (such as those of hydrophobins and hydrophobin-like proteins) play such a paramount role that all other mechanisms become secondary. Such a finding would certainly shift foci in soil organic N research. Moreover, a new tool that has become available, amino acid chirality analysis (Amelung et al. 2006), could be utilized to infer protein age and thus turnover rates.
It may be valuable to use laboratory-based physiological and biochemical information on proteins to identify targets for the study of soil N stabilization. Hydrophobins and hydrophobin-like proteins (SC15, repellents, chaplins) are structural proteins with very specific functions at interface that clearly merit further study. Glomalin(s) and hydrophobin-like proteins, with a strong potential role in soil aggregate formation and stabilization, also deserve attention. Other potentially important protein groups include any that are highly hydrophobic or toxic, and or even microbial cell-wall proteins. Clearly an increased dialogue between microbial physiologists, protein biochemists and soil scientists would speed progress.
References
Alexandrescu AT (2005) Amyloid accomplices and enforcers. Protein Sci 14:1–12
Anderson PA (1985) Interactions between proteins and constituents that affect protein quality. In: Finley GW, Hopkins DT (eds) Digestibility and amino acid availability in cereals and oilseeds. Am. Assoc. Cerea.l Chem., St Paul, pp 31–46
Amelung W, Zhang X, Flach KW (2006) Amino acids in grassland soils: climatic effects on concentrations and chirality. Geoderma 130:207–217
Angers DA, Recous S, Aita C (1997) Fate of carbon and nitrogen in water-stable aggregates during decomposition of 13C15N-labelled wheat straw in situ. Eur J Soil Sci 48:295–300
Arfaioli P, Pantani OL, Bosseto M, Ristori GG (1999) Influence of clay minerals and exchangeable cations on the formation of humic-like substances (melanoidins) from d-glucose and l-tyrosine. Clay Minerals 34:487–497
Basaraba J, Starkey RL (1966) Effects of plant tannins on decomposition of organic substances. Soil Sci 101:17–23
Bashan Y, Levanony H (1988) Active attachment of Azospirillum-brasilense Cd to quartz sand and to a light-textured soil by protein bridging. J Gen Microbiol 134:2269–2279
Benoit RE, Starkey RL, Basaraba J (1968) Effect of purified plant tannins on decomposition of some organic compounds and plant materials. Soil Sci 105:153–158
Bernard BA, Newton SA, Olden K (1983) Effect of size and location of the oligosaccharide chain on protease degradation of bovine pancreatic ribonuclease. J Biol Chem 258:12198–12202
Blackwood CB, Dell CJ, Smucker AJM, Paul EA (2006) Eubacterial communities in different soil macroaggregate environments and cropping systems. Soil Biol Biochem 38:720–728
Blum U, Rice EL (1969) Inhibition of symbiotic nitrogen-fixation by gallic and tannic acid, and possible roles in old-field succession. Bull Torrey Bot Club 96:531–544
Böckelmann U, Szewzyk U, Grohmann E (2003) A new enzymatic method for the detachment of particle associated soil bacteria. J Microb Meth 55:201–211
Bosetto M, Arfaioli P, Pantani OL (2002) Study of the Maillard reaction products formed by glycine and d-glucose on different mineral substrates. Clay Minerals 37:195–204
Bradford MM (1976) A rapid and sensitive method for quantification of microgram quantities of protein utilizing the principle of protein-dye-binding. Anal Biochem 72:248–254
Bradley R, Titus BD, Preston CM (2000) Changes to mineral N cycling and microbial communities in black spruce humus after addition of (NH4)2SO4 and condensed tannins extracted from Kalmia augustifolia and balsam fir. Soil Biol Biochem 32:1227–1240
Bremner JM (1965) Organic forms of nitrogen. Agronomy 9:1238–1255
Brewer R (1964) Fabric and mineral analysis of soils. Wiley, NY
Brisou JF (1995) Biofilms, methods for enzymatic release of microorganisms. CRC Press. Boca Raton, FL
Burdon J (2001) Are the traditional concepts of the structures of humic substances realistic? Soil Sci 166:752–769
Burns RG (1978) Soil enzymes. Academic Press, New York
Burns RG, Dick RP (2002) Enzymes in the environment: activity, ecology, and applications. Marcel Dekker, New York
Busto MD, Perez-Mateos M (1995) Extraction of humic-β-glucosidase fractions from soils. Biol Fertil Soils 20:77–82
Butko P, Buford JP, Goodwin JS, Stroud PA, McCormick CL, Cannon GC (2001) Spectroscopic evidence for amyloid-like interfacial self-assembly of hydrophobin SC3. Biochem Biophys Res Commun 280:212–215
Butler MJ, Day AW (1998) Fungal melanins: a review. Can J Microbiol 44:1115–1136
Caldwell BA (2005) Enzyme activities as a component of soil biodiversity: a review. Pedobiologia 49:637–644
Carpenter CE, Mueller RJ, Kazmierczak P, Zhang L, Villalon DK, van Alfen NK (1992) Effect of a virus on accumulation of a tissue specific cell surface protein of the fungus Cryphonectria (Endothia) parasitica. Mol Plant-Microbe Interact 4:55–61
Chenu C, Stotzky G (2002) Interactions between microorganisms and soil particles: an overview. In: Huang PM, Bollag JM, Senesi N (eds) interactions between soil particles and microorganisms. John Wiley and Sons, Chichester, UK, pp 3–40
Cheryan M (1980) Phytic acid interactions in food systems. Crit Rev Food Sci Nutr 13:297–335
Christensen BT (1992) Physical fractionation of soil and organic matter in primary particle size and density separates. Adv Soil Sci 20:1–90
Claessen D (2004) Structural proteins involved in morphological differentiation of streptomycetes. Thesis (Microbiology, University of Groningen, The Netherlands
Claessen D, Rink R, de Jong W, Siebring J, de Vreugd P, Boersma FGH, Dijkhuizen L, Wösten HAB (2003) A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils. Genes Dev 17:1714–1726
Claessen D, Stokroos I, Deelstra HJ, Penninga NA, Bormann C, Salas JA, Dijkhuizen L, Wösten HAB (2004) The formation of the rodlet layer of streptomycetes is the result of the interplay between rodlins and chaplins. Mol Microbiol 53:433–443
Coelho RR, Linares LF, Martin JP (1985) Amino acid distribution in some fungal melanins and of soil humic acids from Brazil. Plant Soil 87:337–346
Corvis Y, Walcarius A, Rink R, Mrabet NT, Rogalska E (2005) Preparing catalytic surfaces for sensing applications by immobilizing enzymes via hydrophobin layers. Anal Chem 77:1622–1630
Cosgrove DJ (1966) The chemistry and biochemistry of inositol polyphosphates. Rev Pure Appl Chem 16:209–224
Dalal RC (1977) Soil organic phosphorus. Adv Agron 29:85–117
de Gryze S, Six J, Brits C, Merckx R (2005) A quantification of short-term macro-aggregate dynamics: influences of wheat residue input and texture. Soil Biol Biochem 37:55–66
de Jong JF (2006) Aerial hyphae of Schizophyllum commune: their function and formation. Thesis, University of Utrecht
de Vocht ML, Reviakine I, Ulrich WP, Bergsma-Schutter W, Wösten HAB, Vogel H, Brisson A, Wessels JGH, Robillard GT (2002) Self-assembly of the hydrophobin SC3 proceeds via two structural intermediates. Prot Sci 11:1199–1205
de Vries OMH, Fekkes MP, Wösten HAB, Wessels JGH (1993) Insoluble hydrophobin complexes in the walls of Schizophyllum commune and other filamentous fungi. Arch Microbiol 159:330–335
Díaz-Zorita M, Perfect E, Grove JH (2002) Disruptive methods for assessing soil structure. Soil Tillage Res 64:3–22
Dobson CM (1999) Protein misfolding, evolution and disease. Trend Biochem Sci 24:329–332
Dormaar JF, Smoliak S, Willms WD (1990) Soil chemical properties during succession from abandoned cropland to native range. J Range Manag 43:260–265
Driver JD, Holben WE, Rillig MC (2005) Characterization of glomalin as a hyphal wall component of arbuscular mycorrhizal fungi. Soil Biol Biochem 37:101–106
Elliott ET, Coleman DC (1988) Let the soil work for us. Ecol Bull 39:23–32
Elliot MA, Karoonuthaisiri N, Huang J, Bibb MJ, Cohen SN, Kao CM, Buttner MJ (2003) The chaplins: a family of hydrophobic cell-surface proteins involved in aerial mycelium formation in Streptomyces coelicolor. Genes Dev 17:1727–1740
Fierer N, Schimel JP, Cates RG, Zou J (2001) Influence of balsam poplar tannin fractions on carbon and nitrogen dynamics in Alaskan taiga floodplain soils. Soil Biol Biochem 33:1827–1839
Friedel JK, Scheller E (2002) Composition of hydrolysable amino acids in soilk organic matter and soil microbial biomass. Soil Biol Biochem 34:315–325
Friedenauer S, Berlet HH (1989) Sensitivity and variability of the Bradford protein assay in the presence of detergents. Anal Biochem 178:263–268
Gadkar V, Rillig MC (2006) The arbuscular mycorrhizal fungal protein glomalin is a putative homolog of heat shock protein 60. FEMS Microbiol Lett 263:93–101
Gallet C, Lebreton P (1995) Evolution of phenolic patterns and associated litters and humus of a mountain forest ecosystem. Soil Biol Biochem 27:157–165
Gebbink MFBG, Claessen D, Bouma B, Dijkhuizen L, Wösten HAB (2005) Amyloids – a functional coat for microorganisms. Nat Rev Microbiol 3:333–341
Gil H, Mata-Segreda JF, Schowen RL (1991) Effect of non-enzymatic glycosylation on reactivity in proteolysis [in Spanish] Acta Cient Venez 42:16–23
Grossman RB, Lynn WC (1967) Gel-like films that may form at the air-water interface in soils. Soil Sci Soc Am Proc 31:259–262
Hagerman AE, Rice ME, Ritchard NT (1998) Mechanisms of protein precipitation for two tannins, pentagalloyl glucose and epicatechin16 (4→ 8) catechin (procyanidin). J Agric Food Chem 46:2590–2595
Hagerman AE, Robbins CT (1987) Implication of soluble tannin–protein complexes for tannin analysis and plant defense mechanisms. J Chem Ecol 13:1243–1258
Harner MJ, Ramsey PW, Rillig MC (2004) Protein accumulation and distribution in floodplain soils and river foam. Ecol Lett 7:829–836
Harris P (1972) Micro-organisms in surface films from soil crumbs. Soil Biol Biochem 4:105–106
Haslam E (1989) Plant polyphenols: vegetable tannins revisited. Cambridge University Press, 230 pp
Hättenschwiler S, Vitousek P (2000) The role of polyphenols in terrestrial ecosystem nutrient cycling. Trends Ecol Evol 15:238–243
Horner JD, Gosz JR, Cates RG (1988) The role of carbon-based plant secondary metabolites in decomposition in terrestrial ecosystems. Am Nat 132:869–883
Howard PJA, Howard DM (1993) Ammonification of complexes prepared from gelatin and aqueous extracts of leaves and freshly-fallen litter of trees on different soil types. Soil Biol Biochem 25:1249–1256
Ho JGS, Kitov PI, Paszkiewicz E, Sadowska J, Bundlet DR, Ng KK-S (2005) Ligand-assisted aggregation of proteins. J Biol Chem 280:31999–32008
Hu P-H, Hatcher PG (2003) New evidence for covalent coupling of peptides to humic acids based on 2D NMR spectroscopy: a means for preservation. Geochim Cosmochim Acta 69:4521–4533
Ikan R (1996) The Maillard reaction. Consequences for the chemical and life sciences. John Wiley & Sons, Chichester, UK
Jahnel JB, Frimmel FH (1995) Enzymatic release of amino acids from different humic substances. Acta Hydrochim Hydrobiol 23:31–35
Jakas A, Horvat \({\hat{\hbox{S}}}\) (2004) The effect of glycation on the chemical and enzymatic stability of the endogenous opioid peptide, leucine-enkephalin, and related fragments. Bioorg Chem 32:516–526
Janssen MI, van Leeuwen MBM, Scholtmeijer K, van Kooten TG, Dijkhuizen L, Wösten HAB (2002) Coating with genetic engineered hydrophobin promotes growth of fibroblasts on a hydrophobic solid. Biomaterials 23:4847–4854
Janssen MI, van Leeuwen MBM, van Kooten TG, de Vries J, Dijkhuizen L and Wösten HAB (2004) Promotion of cell activity by coating with hydrophobin in the β-sheet end state. Biomaterials 25:2731–2739
Jastrow JD (1996) Soil aggregate formation and the accrual of particulate and mineral-associated organic matter. Soil Biol Biochem 28:665–676
Jokic A, Frenkel AI, Vairavamurthy MA, Huang PM (2001) Birnssite catalysis of the Maillard reaction: its significance in natural humification. Geophys Res Lett 28:3899–3902
Jokic A, Wang MC, Liu C, Frenkel AI, Huang PM (2004) Integration of the polyphenol and Maillard reactions into a unified abiotic pathway for humification in nature: the role of δ-MnO2. Org Geochem 35:747–762
Keeney DR, Bremner JM (1964) Effect of cultivation on the nitrogen distribution in soils. Soil Sci Soc Am Proc 28:653–656
Kleber M, Sollins P, Sutton R (2006) A conceptual model of organo-mineral interactions in soils: self assembly of organic molecular fragments into zonal structures on mineral surfaces. Biogeochemistry (this volume)
Knicker H (2006) Vegetation fires and burnings; how do they affect the nature and stability of soil organic nitrogen and carbon? – A review. Biogeochemistry (this volume)
Knicker H (2000) Biogenic Nitrogen in soils as revealed by solid-state Carbon-13 and Nitrogen-15 Nuclear Magnetic Resonance spectroscopy. J Environ Qual 29:715–723
Knicker H, Hatcher PG (1997) Survival of protein in an organic-rich sediment. Possible protection by encapsulation in organic matter. Naturwissenschaften 84:231–234
Knicker H, Schmidt MWI, Kögel-Knabner I (2000) Nature of organic nitrogen in fine particle separates of sandy soils in highly industrialized area as revealed by NMR spectroscopy. Soil Biol Biochem 32:241–252
Kojima RT (1947) Soil organic nitrogen: I. Nature of the organic nitrogen in a muck soil from Geneva, New York. Soil Sci 64:157–165
Kranabetter JM, Banner A (2000) Selected biological and chemical properties of forest floors across bedrock types on the northern coast of British Columbia. Can J For Res 30:971–981
Kraus TEC, Dahlgren RA, Zasoski RJ (2003a) Tannins in nutrient dynamics of forest ecosystems: a review. Plant Soil 256:41–66
Kraus TEC, Yu Z, Preston CM, Dahlgren RA, Zasoski RJ (2003b) Linking chemical reactivity and protein precipitation to structural characteristics of foliar tannins. J Chem Ecol 29:703–730
Kuiters AT, Denneman CAJ (1987) Water-soluble phenolic substances in soils under several coniferous and deciduous tree species. Soil Biol Biochem 19:765–769
Kuo M-J, Alexander M (1967) Inhibition of the lysis of fungi by melanins. J Bacteriol 94:624–629
Ladd JN, Brisbane PG (1967) Release of amino acids from soil humic acids by proteolytic enzymes. Aust J Soil Res 5:161–171
Leinweber P, Schulten H-R (2000) Nonhydrolyzable forms of soil organic nitrogen: extractability and composition. J Plant Nutr Soil Sci 163:433–439
Lewis JA, Starkey RL (1968) Vegetable tannins, their decomposition and effects on decomposition of some organic compounds. Soil Sci 106:241–247
Linder M, Szilvay GR, Nakari-Setälä T, Söderlund H, Penttilä M (2002) Surface adhesion of fusion proteins containing the hydrophobins HFBI and HFBII from Trichoderma reesei. Protein Sci 11:2257–2266
Lipson DA, Schmidt SK, Monson RK (1999) Links between microbial population dynamics and nitrogen availability in an alpine ecosystem. Ecology 80:1623–1631
Lockwood JL (1960) Lysis of mycelia of plant pathogenic fungi by natural soil. Phytopathology 50:787–789
Loll MJ, Bollag J-M (1983) Protein transformation in soil. Adv Agron 36:351–382
Loomis WD, Battaile J (1966) Plant phenolic compounds and the isolation of plant enzymes. Phytochemistry 5:423–438
Lorenz K, Preston CM (2002) Characterization of high-tannin fractions from humus by carbon-13 cross-polarization and magic angle spinning nuclear magnetic resonance. J Environ Qual 31:431–436
Lorenz K, Preston CM, Raspe S, Morrison IK, Feger KH (2000) Litter composition and humus characteristics in Canadian and German spruce ecosystems: information from tannin analysis and 13C CPMAS NMR. Soil Biol Biochem 32:779–792
Lovelock CE, Wright SF, Clark DA, Reuss RW (2004) Soil stocks of glomalin produced by arbuscular mycorrhizal fungi across a tropical rain forest landscape. J Ecol 92:278–287
Lugones LG, Bosscher JS, Scholtmeijer K, de Vries OMH, Wessels JGH (1996) An abundant hydrophobin (ABH1) forms hydrophobic rodlet layers in Agaricus bisporus fruiting bodies. Microbiology 142:1321–1329
Lugones LG, Wösten HAB, Wessels JGH (1998) A hydrophobin (ABH3) specifically secreted by vegetatively growing hyphae of Agaricus bisporus (common white button mushroom). Microbiology 144:2345–2353
Lugones LG, Wösten HAB, Birkenkamp KU, Sjollema KA, Zagers J, Wessels JGH (1999) Hydrophobins line air channels in fruiting bodies of Schizophyllum commune and Agaricus bisporus. Mycol Res 103:635–640
Lugones LG, de Jong JF, de Vries OMH, Jalving R, Dijksterhuis J, Wösten HAB (2004) The SC15 protein of Schizophyllum commune mediates formation of aerial hyphae and attachment in the absence of the SC3 hydrophobin. Mol Microbiol 53:707–716
Lutgen ER, Clairmont DL, Graham J, Rillig MC (2003) Seasonality of arbuscular mycorrhizal hyphae and glomalin in a western Montana grassland. Plant Soil 257:71–83
Martens DA, Reedy TE, Lewis DT (2003) Soil organic carbon content and composition of 130-year crop, pasture and forest land-use managements. Global Change Biol 10:65–78
Martens DA, Loeffelmann KL (2003) Soil amino acid composition quantified by acid hydrolysis and anion-chromatography-pulsed amperometry. J Agric Food Chem 51:6521–6529
Martin JP, Parsa AA, Haider K (1978) Influence of intimate association with humic polymers on biodegradation of [14C]labeled organic substrates in soil. Soil Biol Biochem 10:483–486
Matsumoto S, Ae N, Yamagata M (2000) Extraction of mineralizable organic nitrogen from soils by a neutral phosphate buffer solution. Soil Biol Biochem 32:1293–1299
McManus JP, Davis KG, Lilley TH, Haslam E (1981) The association of proteins with polyphenols. J Chem Soc Commun 309–311
Merlini G, Belloti V (2003) Molecular mechanisms of amyloidosis. N Engl J Med 349:583–596
Miltner A, Zech W (1999) Microbial degradation and resynthesis of protein during incubation of beech leaf litter in the presence of mineral phases. Biol Fertil Soils 30:48–51
Mummey DL, Holben W, Six J, Stahl P (2006) Spatial stratification of soil bacterial populations in aggregates of diverse soils. Microb Ecol 51:404–411
Nakas JP, Klein DA (1979) Decomposition of microbial cell components in a semi-arid grassland soil. Appl Environ Microbiol 38:454–460
Németh K, Bartels H, Vogel M, Mengel K (1988) Organic nitrogen compounds extracted from arable and forest soils by electro-ultrafiltration and recovery rates of amino acids. Biol Fertil Soils 5:271–275
Nierop KGJ, Verstaten JM, Tietma A, Westervald JW, Wartenbergh PE (2006) Short- and long-term tannin induced carbon, nitrogen and phosphorus dynamics in Corsican pine litter. Biogeochemistry 79:275–296
Northup RR, Yu Z, Dahlgren RA, Vogt KA (1995) Polyphenol control of nitrogen release from pine litter. Nature 377: 227–229
Northup RR, Dahlgren DA, McColl JG (1998) Polyphenols as regulators of plant–litter–soil interactions in northern California’s pygmy forest: A positive feedback? Biogeochemistry 42:189–220
Nyman BF (1985) Protein-proanthocyanidin interactions during extraction of Scots pine needles. Phytochemistry 24:2939–2944
Oades JM (1984) Soil organic matter and structural stability: mechanisms and implications for management. Plant Soil 76:319–337
Oh HI, Hoff JE, Armstrong GS, Haff LA (1980) Hydrophobic interactions in tannin–protein complexes. J Agric Food Chem 28:394–398
Omoike A, Chorover J (2004) Spectroscopic study of extracellular polymeric substances from Bacillus subtilis: aqueous chemistry and adsorption effects. Biomacromolecules 5:1219–1230
Opdenakker G, Rudd PM, Ponting CP, Dwek RA (1993) Concepts and principles pf glycobiology. FASEB J 7:1330–1337
Parsek MR, Fuqua C (2004) Biofilms 2003: emerging themes and challenges in studies of surface-associated microbial life. J Bacteriol 186:4427–4440
Paul EA, Clark FE (1996) Soil microbiology and biochemistry, 2nd edn. Academic Press, Orlando
Piccolo A (2001) The supramolecular structure of humic substances. Soil Sci 166:810–832
Plante A, McGill W (2002) Soil aggregate dynamics and the retention of organic matter in laboratory-incubated soil with differing simulated tillage frequencies. Soil Tillage Res 66:79–92
Plante AF, Feng Y, McGill WB (2002) A modeling approach to quantifying soil macroaggregate dynamics. Can J Soil Sci 82:181–190
Raab TK, Lipson DA, Monson RK (1999) Soil amino acid utilization among species of the Cyperaceae: plant and soil processes. Ecology 80:2408–2419
Ravindran V, Bryden WL, Kornegay ET (1995) Phytates: occurrence, bioavailability and implications in poultry nutrition. Poult Avian Bio Rev 6:125–143
Rice EL, Pancholy SK (1973) Inhibition of nitrification by climax ecosystems II Additional evidence and possible role of tannins. Am J Bot 60:691–702
Rillig MC (2004) Arbuscular mycorrhizae, glomalin and soil quality. Can J Soil Sci 84:355–363
Rillig MC (2005) A connection between fungal hydrophobins and soil water repellency. Pedobiologia 49:395–399
Rillig MC, Mummey DL (2006) Mycorrhizas and soil structure. New Phytol 171:41–53
Rillig MC, Wright SF, Allen MF, Field CB (1999) Rise in carbon dioxide changes soil structure. Nature 400:628–628
Rillig MC, Wright SF, Nichols KA, Schmidt WF, Torn MS (2001) Large contribution of arbuscular mycorrhizal fungi to soil carbon pools in tropical forest soils. Plant Soil 233:167–177
Rillig MC, Wright SF, Eviner VT (2002) The role of arbuscular mycorrhizal fungi and glomalin in soil aggregation: comparing effects of five plant species. Plant Soil 238:325–333
Rillig MC, Ramsey PW, Morris S, Paul EA (2003) Glomalin, an arbuscular-mycorrhizal fungal soil protein, responds to land-use change. Plant Soil 253:293–299
Rosier CL, Hoye A, Rillig MC (2006) Glomalin-related soil protein: assessment of current detection and quantification tools. Soil Biol Biochem 38:2205–2211
Russo PS, Blum FD, Ipsen JD, Miller WG, Abul-Hajj YJ (1982) The surface activity of the phytotoxin cerato-ulmin. Can J Bot 60:1414–1422
Sarkar JM, Burns RG (1984) Synthesis and properties of β-glucosidase-phenolic copolymers as analogues of soil humic-enzyme complexes. Soil Biol Biochem 16:619–625
Schlesinger WH (1991) Biogeochemistry – an analysis of global change. Academic Press, San Diego
Schofield JA, Hagerman AE, Harold A (1998) Loss of tannins and other phenolics from willow litter. J Chem Ecol 24:1409–1421
Scholtmeijer K, Janssen MI, Gerssen B, de Vocht ML, van Leeuwen MBM, van Kooten TG, Wösten HAB, Wessels JGH (2002) Surface modification created by using engineered hydrophobins. Appl Environ Microbiol 68:1367–1373
Schulten H-R, Schnitzer M (1993) A state of the art structural concept for humic substances. Naturwissenschaften 80:29–30
Schulze WX (2005) Protein analysis in dissolved organic matter: what proteins from organic debris, soil leachate and surface water can tell us – a perspective. Biogeosciences 2:75–86
Schulze WX, Gleixner G, Kaiser K, Guggenberger G, Mann M, Schulze ED (2005) A proteomic fingerprint of dissolved organic carbon and of soil particles. Oecologia 142:335–343
Sen R (2003) The root-microbe-soil interface: new tools for sustainable plant production. New Phytol 157:391–393
Sexstone AJ, Revsbech NP, Parkin TB, Tiedje JM (1985) Direct measurement of oxygen profiles and denitrification rates in soil aggregates. Soil Sci Soc Am J 49:645–651
Simonart P, Batistic L, Mayaudon J (1967) Isolation of protein from humic acid extracted from soil. Plant Soil 27:153–161
Singh M, Krikorian AD (1982) Inhibition of trypsin activity in vitro by phytate. J Agric Food Chem 30:799–800
Six J, Feller C, Denef K, Ogle SM, de Moraes JC, Albrecht A (2002) Soil organic matter, biota and aggregation in temperate and tropical soils – effects of no-tillage. Agronomie 22:755–775
Six J, Bossuyt H, Degryze S, Denef K (2004) A history of research on the link between (micro)aggregates, soil biota, and soil organic matter dynamics. Soil Tillage Res 79:7–31
Six J, Elliott ET, Paustian K, Doran JW (1998) Aggregation and soil organic matter accumulation in cultivated and native grassland soils. Soil Sci Soc Am J 62:1367–1377
Smernik RJ, Baldock JA (2005) Does solid-state 15N NMR spectroscopy detect all soil organic nitrogen? Biogeochemistry 75:507–528
Smolander A, Loponen J, Suominen K, Kitunen V (2005) Organic matter characteristics and C and N transformations in the humus layer under two tree species, Betula pendula and Picea abies. Soil Biol Biochem 37:1309–1318
Sollins P, Homan P, Caldwell BA (1996) Stabilization and destabilization of soil organic matter: mechanisms and controls. Geoderma 74:65–105
Sommerville K, Preston T (2001) Characterization of dissolved combined amino acids in marine waters. Rapid Commun Mass Spectrom 15:1287–1290
Sørensen LH (1975) The influence of clay on the rate of decay of amino acid metabolites synthesized in soils during decomposition of cellulose. Soil Biol Biochem 7:171–177
Sowden FJ, Chen Y, Schnitzer M (1977). The nitrogen distribution in soils formed under widely differing climatic conditions. Geochim Cosmochim Acta 41:1524–1526
Steinberg PD, Rillig MC (2003) Differential decomposition of arbuscular mycorrhizal fungal hyphae and glomalin. Soil Biol Biochem 35:191–194
Sternberger RE, Holden PA (2004) Macromolecular composition of unsaturated Psuedomonas aeruginosa biofilms with time and carbon source. Biofilms 1:37–47
Stevenson FJ (1994) Humus chemistry: genesis, composition, reactions. Wiley Interscience, New York
Suominen K, Kitunen V, Smolander A (2003) Characteristics of dissolved organic matter and phenolic compounds in forest soils under silver birch (Betula pendula), Norway spruce (Picea abies) and Scots pine (Pinus sylvestris). Eur J Soil Sci 54:287–293
Sutton R, Sposito G (2005) Molecular structure in soil humic substances: the new view. Environ Sci Technol 39:9009–9015
Takai S, Richards WC (1978) Cerato-ulmin, a wilting toxin of Ceratocystis ulmi: isolation and some properties of cerato-ulmin from the culture of C. ulmi Phytopathol Z 91:129–146
Theng BKG (1979) Formation and properties of clay-polymer complexes. Elsevier Scientific Pub C, New York
Tisdall JM, Oades JM (1982) Organic matter and water-stable aggregates in soils J Soil Sci 33:141–163
Torkkeli M, Serimaa R, Ikkala O, Linder M (2002) Aggregation and self-assembly of hydrophobins from Trichoderma reesei: low-resolution structural models. Biophys J 83:2240–2247
Varki A (1993) Biological roles of oligosaccharides: all of the theories are correct. Glycobiology 3:97–130
Verma L, Martin JP, Haider K (1975) Decomposition of carbon-14-labeled proteins, peptides, and amino acids; free and complexed with humic polymers. Soil Sci Soc Am Proc 39:279–284
Wang X, Shi F, Wösten HAB, Hektor H, Poolman B, Robillard GT (2005) The SC3 hydrophobin self-assembles into a membrane with distinct mass transfer properties. Biophys J 88:3434–3443
Waksman SA (1938) Humus. Williams & Wilkins, Baltimore
Waksman SA, Iyer KRN (1932) Contribution to our knowledge of the chemical nature and origin of humus: I On the synthesis of the “humus nucleus”. Soil Sci 34:43–69
Weintraub MN, Schimel JP (2005) Seasonal protein dynamics in Alaskan arctic tundra soils. Soil Biol Biochem 37:1469–1475
Wessels JGH (1994) Developmental regulation of fungal cell wall formation. Ann Rev Phytopathol 32:413–437
Wessels JGH,Ásgeirsdóttir SA, Birkenkamp KU, de Vries OMH, Lugones LG, Scheer JMJ, Schuren FHJ, Schuurs TA, van Wetter M-A, Wösten HAB (1995) Genetic regulation of emergent growth in Schizophyllum commune. Can J Bot 73: S273–S281
Wessels JGH, de Vries OMH, Ásgeirsdóttir SA, Schuren FHJ (1991a) Hydrophobin genes involved in formation of aerial hyphae and fruit bodies in Schizophyllum. Plant Cell 3:793–799
Wessels JGH, de Vries OMH, Ásgeirsdóttir SA, Springer J (1991b) The thn mutation of Schizophyllum commune which suppresses formation of aerial hyphae affects expression of the SC3 hydrophobin gene. J Gen Microbiol 137:2439–2445
West CM (1986) Current ideas on the significance of protein glycosylation. Mol Cell Biochem 72:3–20
Whiteford JR, Spanu PD (2002) Hydrophobins and the interactions between fungi and plants. Mol Plant Pathol 3:391–400
Wösten HAB (2001) Hydrophobins: multipurpose proteins. Ann Rev Microbiol 55:625–646
Wösten HAB, de Vocht ML (2000) Hydrophobins, the fungal coat unravelled. Biochim Biophys Acta 1469:79–86
Wösten HAB, de Vries OMH, Wessels JGH (1993) Interfacial self-assembly of a fungal hydrophobin into a hydrophobic rodlet layer. Plant Cell 5:1567–1574
Wösten HAB, Ruardy TG, van der Mei HC, Busscher HJ, Wessels JGH (1995) Interfacial self-assembly of a Schizophyllum commune hydrophobin into an insoluble amphipathic protein membrane depends on surface hydrophobicity. Colloids Surf B: Biointerfaces 5:189–195
Wösten HAB, Schuren FHJ, Wessels JGH (1994) Interfacial self-assembly of a hydrophobin into an amphipathic protein membrane mediates fungal attachment to hydrophobic surfaces. EMBO J 13: 5848–5854
Wösten HAB, Bohlmann R, Eckerskorn C, Lottspeich F, Bölker M, Kahmann R (1996) A novel class of small amphipathic peptides affect aerial hyphal growth and surface hydrophobicity in Ustilago maydis. EMBO J 15:4274–4281
Wösten HAB, van Wetter M-A, Lugones LG, van der Mei HC, Busscher HJ, Wessels JGH (1999) How a fungus escapes the water to grow into the air. Curr Biol 9:85–88
Wright SF, Anderson RL (2000) Aggregate stability and glomalin in alternative crop rotations for the central Great Plains. Biol Fertil Soils 31:249–253
Wright SF, Upadhyaya A (1998) A survey of soils for aggregate stability and glomalin, a glycoprotein produced by hyphae of arbuscular mycorrhizal fungi. Plant Soil 198:97–107
Wright SF, Upadhyaya A (1996) Extraction of an abundant and unusual protein from soil and comparison with hyphal protein of arbuscular mycorrhizal fungi. Soil Sci 161:575–586
Yu Z, Zhang Q, Kraus TEC, Dahlgren RA, Anastacio C, Zasoski RJ (2002) Contribution of amino compounds to dissolved organic nitrogen in forest soils. Biogeochemistry 61:173–198
Zang X., Van Heemst J, Jasper DH, Dria KJ, Hatcher PG (2000) Encapsulation of protein in humic acid from Histosols as an explanation for the occurrence of organic nitrogen in soil and sediment. Org Geochem 31:679–695
Zucker WV (1983) Tannins: does structure determine function ? An ecological perspective. Am Nat 121:335–365
Acknowledgements
Funding for the Asilomar Conference on Mechanisms of SOM Stabilization, which brought the authors together for the first time, was provided by NSF, USDA-NRI, Kearney Foundation of Soil Science, Livermore National Laboratory, NASA, and the Forest Science Department, Oregon State University. MCR, BAC and PS acknowledge funding by the NSF Ecosystem Studies program and USDA NRI CSREES. We thank Dr. D. Mummey for comments on an earlier version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rillig, M.C., Caldwell, B.A., Wösten, H.A.B. et al. Role of proteins in soil carbon and nitrogen storage: controls on persistence. Biogeochemistry 85, 25–44 (2007). https://doi.org/10.1007/s10533-007-9102-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10533-007-9102-6