
Abstract
Two aerobic, lab-scale, slurry-phase bioreactors were used to examine the biodegradation of polycyclic aromatic hydrocarbons (PAHs) in contaminated soil and the associated bacterial communities. The two bioreactors were operated under semi-continuous (draw-and-fill) conditions at a residence time of 35 days, but one was fed weekly and the other monthly. Most of the quantified PAHs, including high-molecular-weight compounds, were removed to a greater extent in the weekly-fed bioreactor, which achieved total PAH removal of 76%. Molecular analyses, including pyrosequencing of 16S rRNA genes, revealed significant shifts in the soil bacterial communities after introduction to the bioreactors and differences in the abundance and types of bacteria in each of the bioreactors. The weekly-fed bioreactor displayed a more stable bacterial community with gradual changes over time, whereas the monthly-fed bioreactor community was less consistent and may have been more strongly influenced by the influx of untreated soil during feeding. Phylogenetic groups containing known PAH-degrading bacteria previously identified through stable-isotope probing of the untreated soil were differentially affected by bioreactor conditions. Sequences from members of the Acidovorax and Sphingomonas genera, as well as the uncultivated “Pyrene Group 2” were abundant in the bioreactors. However, the relative abundances of sequences from the Pseudomonas, Sphingobium, and Pseudoxanthomonas genera, as well as from a group of unclassified anthracene degraders, were much lower in the bioreactors compared to the untreated soil.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
There are an estimated 45,000 former manufactured gas plant (MGP) and related coal tar sites in the United States, remnants from gas production in the nineteenth and early twentieth centuries (U.S. Environmental Protection Agency 2004). Soils at many of these sites remain heavily contaminated with a variety of hazardous compounds, including polycyclic aromatic hydrocarbons (PAHs) that were components of both the fossil fuels used to generate manufactured gas and byproducts of the process (Haeseler et al. 1999). PAHs remain a concern at these sites, as they can persist in soils for prolonged periods of time due to their high hydrophobicity (Cerniglia 1992). Of this broad class of chemicals, 16 PAHs are regulated by the United States Environmental Protection Agency (EPA) as priority pollutants, and many of the higher-molecular-weight (HMW) PAHs, such as benzo[a]pyrene, are considered human carcinogens (Agency for Toxic Substances and Disease Registry 2007; U.S. Environmental Protection Agency 1993).
Remediation of MGP sites and other PAH-impacted soils can be cost intensive (U.S. Environmental Protection Agency 2004). A variety of methods such as incineration, soil vapor extraction, thermal desorption, and bioremediation are often employed for site cleanup (U.S. Environmental Protection Agency 2004, 2007). Although an attractive option for many reasons, bioremediation often results in incomplete removal of PAHs, with the largest fraction remaining generally composed of the more recalcitrant and hazardous HMW compounds (Lundstedt et al. 2003; Ringelberg et al. 2001). There is little published information on how variables associated with engineered biological treatment systems influence both PAH removal from contaminated soil and the relevant microbial communities in those systems. On the assumption that PAH removal depends at least in part on the composition of the microbial community, it is important to begin characterizing these communities with emerging molecular tools.
A wide range of microorganisms have the capability to degrade one or more PAHs. The lower-molecular-weight (LMW) PAHs, such as naphthalene and phenanthrene, are commonly used as growth substrates by members of diverse bacterial genera. Some HMW compounds, such as pyrene, are also capable of supporting microbial growth, particularly by Gram-type positive bacteria (Dean-Ross and Cerniglia 1996; Khan et al. 2002). However, recent studies utilizing stable isotope probing (SIP) have revealed groups of uncultivated Proteobacteria that also play a role in pyrene removal (Jones et al. 2008; Singleton et al. 2006). PAHs of five or more rings have not been found to support bacterial growth and are generally transformed through co-metabolism by organisms that grow on some other substrate (Kanaly and Harayama 2000). Knowledge of the organisms capable of transforming specific PAHs and how they respond to particular environmental stimuli may help in the design, improvement, and implementation of future bioremediation efforts.
We studied the biological removal of PAHs from a weathered, contaminated soil excavated from a former MGP site. Two lab-scale, aerobic bioreactors were employed to examine the effects of different feeding patterns on PAH removal. Both reactors were operated semi-continuously at the same effective residence time but with different draw-and-fill cycles, which we hypothesized might lead to different PAH-degrading communities and corresponding differences in PAH removal. Barcoded pyrosequencing of 16S rRNA genes from bioreactor samples and untreated soil was used to determine the dominant members of the communities selected in each reactor.
Methods and materials
Soil
PAH-contaminated soil was obtained from the site of a former MGP located in Salisbury, North Carolina, USA and was air-dried, screened through 10 mm wire mesh, and blended at the time of sampling before storing at 4°C. The soil contained 64% sand, 30% silt and 6% clay, with total organic matter of 8.6% as determined by a thermogravimetric method (Lukasewycz and Burkhard 2005) and extractable organic matter of 14 mg per g of dry soil. Prior to introduction to the bioreactors, a sample of stored soil of sufficient mass for the entirety of the study was additionally screened through a 3.35 mm wire-mesh screen and blended before being returned to storage at 4°C. The moisture content of this soil was approximately 15%. PAH concentrations are provided below.
Bioreactor operation
Two physically identical bioreactors were used, whose general design is described elsewhere (Singleton et al. 2005; Zhu et al. 2008). The reactors were started by adding 2.5 l per bioreactor of a nominal 10% slurry (w/v) of soil in a buffer containing 2.5 mM phosphate and 2.5 mM NH4NO3 (pH 7.5). During feeding of the bioreactors, slurry was filtered through a 2-mm wire mesh screen to remove small rocks and other debris. Both bioreactors were operated aerobically in batch mode for 50 days with constant mixing and aeration. On days 14 and 45 of this startup period, one-half of the volume of each reactor was removed and exchanged into the other reactor to ensure that the microbial communities would be as similar as possible prior to starting semi-continuous operation.
After the batch startup period, the bioreactors were operated as semi-continuous (draw-and-fill) aerobic reactors for 140 days. The first day on which each reactor was fed after the startup period was defined as day 0. During semi-continuous operation the volume in each reactor was reduced to 2 l and the buffer recipe was modified to 5 mM phosphate and 5 mM NH4NO3 (pH 7.5). For one reactor, a total of 80% of the slurry (1.6 l) was removed and replaced with slurried, untreated soil every 28 days (this bioreactor was designated the “monthly-fed reactor”). In the second reactor, 20% of the slurry (0.4 l) was removed and replaced every 7 days (designated the “weekly-fed bioreactor”), establishing an effective residence time of 35 days in each bioreactor. The contents of the bioreactors were continuously aerated and mixed.
Slurry effluent which had been treated in the bioreactors and removed immediately prior to each feeding event was used to quantify PAH concentrations. PAHs in the weekly-fed bioreactor were quantified weekly for the first 3 months of semi-continuous operation and monthly thereafter. The PAH concentrations in feed (untreated) soil were analyzed at days 0, 56 and 140. Data from the day 0 and day 56 feed soil samples were averaged for comparisons to the bioreactor samples.
To test the effects of nitrogen concentration on the pH of slurry, 50-ml volumes of 10% soil slurry (w/v) were created in 250-ml glass flasks and closed with foam stoppers to allow gas exchange. The slurry comprised either 80 or 20% of contents from the weekly-fed bioreactor with the remainder of slurry freshly created from stored, untreated soil. The concentration of phosphate in each newly created slurry buffer was maintained at 5 mM for all flasks. In two sets of control flasks (one set each simulating either weekly-fed or monthly-fed reactor conditions), the nitrogen concentration was identical to the typical bioreactor feeding conditions (5 mM). In a third set of samples simulating the weekly-fed reactor conditions but at an increased nitrogen load, the final concentration of NH4NO3 in the buffer was adjusted to 20 mM. In a fourth set of samples simulating the monthly-fed reactor conditions but at a reduced nitrogen load, the final concentration of NH4NO3 in the buffer was adjusted to 1.25 mM. Triplicate samples were used for each condition and were incubated on a shaker at 225 rpm at 25°C.
Chemical analyses
The pH of slurry was monitored using an Expandable Ion Analyzer EA920 meter (Orion Research, Boston, MA, USA). Triplicate 200-ml effluent slurry samples from each bioreactor at each sampling event were centrifuged, PAHs extracted, and the PAH concentrations determined by high-performance liquid chromatography (HPLC) as previously described (Singleton et al. 2008); the HPLC method was able to quantify 14 of the 16 EPA priority pollutant PAHs. Triplicate samples of stored, untreated feed soil were also slurried in 200 ml reactor buffer and the PAHs quantified by HPLC. Statistical analyses of PAH data were carried out with ProStat version 4.02 (Poly Software International, Pearl River, NY).
Molecular analyses
Total community DNA was obtained from effluent slurry from the bioreactors for the same samples described above for PAH quantification, and from feed soil samples at days 0 and 140. DNA was extracted using a FastDNA® Spin Kit for Soil (MP Biomedicals, Solon, OH, USA) and stored at −20°C in Tris–EDTA buffer (TE; pH 8.0). Denaturing gradient gel electrophoresis (DGGE) was performed as previously described (Singleton et al. 2008), except that the denaturant concentrations of the gel ranged from 40 to 60%, and a non-denaturing stacking gel was employed.
Barcoded 16S rRNA gene pyrosequencing was performed to analyze the bacterial communities of the untreated feed soil and the bioreactors. Ten-fold dilutions of extracted DNA samples in water were used as template for triplicate PCR reactions for each bioreactor at each of the monthly sampling time-points, as well as the feed soil from the beginning (day 0) and end (day 140) of the experiment. Primer pairs 27f and 338r were modified to incorporate an identical 8-base-pair (bp) barcode sequence unique to each sample and a 2-bp spacer on the 5′ end of the primer sequence (Hamady et al. 2008). Each 20 μl PCR reaction was run for 25 cycles of 94°C for 45 s, 55°C for 45 s, and 72°C for 1 min on an Eppendorf (Westbury, NY, USA) Mastercycler Gradient thermal cycler before verification of the proper amplicon size on a 1% agarose gel. The triplicate reactions for each sample were pooled and purified with a QIAquick PCR Purification Kit (Qiagen, Valencia, CA, USA) and eluted in 30 μl of 10 mM Tris–Cl (pH 8.5) buffer. The DNA concentration of pooled amplicons was then measured using a NanoDrop ND-3300 Fluorospectrometer (Thermo, Waltham, MA, USA) and Quant-iT Picogreen dsDNA Kit (Invitrogen, Carlsbad, CA, USA) prior to combining into a single sample at a concentration suitable for pyrosequencing. The sample was submitted to the High-Throughput Sequencing Facility at the University of North Carolina-Chapel Hill for sequencing using the 454 Life Sciences Titanium platform (Roche Diagnostics, Branford, CT, USA).
Segregation of the barcoded sequences into libraries was performed using the Pyro pipeline software of the Ribosome Database Project-II (RDP-II), and alignments, clustering, and dereplication were provided by the same (Cole et al. 2009). After trimming of the barcodes and primers, any sequence less than 250 bp or containing ambiguous bases was removed from further analyses. Phylogenetic assignments for sequences were determined using the Classifier program of the RDP-II with an 80% confidence value threshold (Wang et al. 2007). The number of sequences associated with PAH-degrading phylogenetic groups unrecognized by the RDP-II was determined by BLAST+ 2.2.23 searches against the pyrosequence libraries with representative sequence(s) from each group as a query. Pyrosequences with ≥97% sequence similarity over at least 250 bp to the query sequence were considered a putative member of that group. GenBank accession numbers DQ123671 and HM596265 were used as the query sequences for “Pyrene Group 2” and “Anthracene Group 1,” respectively. These, and other bacterial groups identified as containing PAH-degraders were previously determined by stable-isotope probing (Jones 2010). Briefly, the untreated feed soil from the Salisbury MGP site was batch incubated as a slurry in flasks containing uniformly labeled 13C-naphthalene, phenanthrene, anthracene, fluoranthene, benz[a]anthracene, or pyrene. After incubation to a determined endpoint, DNA was extracted from the soil, separated by ultracentrifugation, and the bacterial 16S rRNA genes present in 13C-enriched DNA fractions identified by molecular methods.
An alignment for phylogenetic tree construction to perform community comparisons was generated using MUSCLE (Edgar 2004) with representative sequences from each library representing clusters at 97% sequence similarity. The alignment was then trimmed using the gBlocks software (Talavera and Castresana 2007) and the tree constructed using the FastTree algorithm (Price et al. 2009). Community comparisons were performed using the Fast UniFrac program with abundance weights (Hamady et al. 2010). Data from pyrosequencing reactions were submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (Shumway et al. 2010) under the study accession number SRP002354.
Results
Bioreactor pH
Prior to introduction to each bioreactor, untreated soil was slurried using a phosphate buffer to a pH of approximately 7.5. At the end of the 50-day batch startup period, the pH values of the slurry in what would become the weekly- and monthly-fed bioreactors were 7.4 and 7.2, respectively. However, subsequent to the initiation of semi-continuous operation the pH in each bioreactor dropped significantly, and the pH of the monthly-fed bioreactor dropped more dramatically and was consistently lower than in the weekly-fed bioreactor throughout the course of the study (Fig. 1). The average measured pH for the weekly-fed bioreactor during the experimental period was 7.0 ± 0.2, compared to 6.4 ± 0.1 for the monthly-fed bioreactor, although the weekly-fed bioreactor displayed a trend of decreasing pH through approximately the first 60 days.

Measured pH of the bioreactor slurries during semi-continuous operation. Open circles represent samples from the monthly-fed bioreactor and closed circles are from the weekly-fed bioreactor
To evaluate whether nitrification of the ammonium added to the bioreactors in the buffer might have caused the observed pH discrepancy, batch incubations simulating the two feeding patterns but with varying amounts of added NH4NO3 were conducted. As instantaneous nitrogen loading was higher in the monthly-fed bioreactor (approximately 600 mg of NH4NO3 per feeding) than in the weekly-fed reactor (150 mg per feeding), the incubations were designed to test the effects of higher nitrogen loading under a simulated weekly-fed condition and lower nitrogen loading under a simulated monthly-fed condition (Fig. 2). Incubations containing higher levels of nitrogen displayed correspondingly lower pH values, while the opposite phenomenon was observed for incubations containing lower levels of nitrogen. In a separate experiment in which slurry samples from each bioreactor were centrifuged and the supernatant removed and replaced with fresh buffer containing the same nitrogen concentration (5 mM) for each sample, the final pH was the same regardless of the source of the slurry (data not shown). These data suggest that the difference in pH between the bioreactors was associated with instantaneous nitrogen loading rather than differences in the nitrification potential of the microbial communities in the reactors.

Measured pH of batch slurry incubations with varying levels of nitrogen amendment. Circles indicate normal nitrogen loading, identical to bioreactor conditions, with open circles representing a monthly-fed condition and closed circles a weekly-fed condition. Squares indicate a modified nitrogen load. The open squares represent a monthly-fed condition with decreased nitrogen loading and closed squares represent a weekly-fed condition with increased nitrogen loading. Data points and error bars are the average and standard deviation, respectively, of triplicate incubations
Bioreactor PAH removal
During the feeding event at day 112 after the beginning of semi-continuous operation (month 4), a blockage in the air delivery system in the monthly-fed bioreactor was discovered and corrected. Therefore, direct comparisons between the two bioreactors for PAH removal were limited to data from the first 3 months of semi-continuous operation (days 28, 56, and 84). Samples from the weekly-fed bioreactor extracted concurrently with the monthly feeding events were used for comparisons. Student’s t-tests indicated that the mean PAH concentrations of weekly-fed bioreactor slurry at these points were not significantly different from the mean concentrations from all weekly sampling events to that point (P < 0.05).
Both bioreactors contained significantly lower concentrations of quantified PAHs compared to the feed soil (P < 0.05) except for naphthalene, which was significant for the weekly-fed bioreactor only, and benzo[g,h,i]perylene (BgP), which was not significantly reduced in either bioreactor (Fig. 3). Compared to mean concentrations in the feed soil, an average of 76 ± 1 and 67 ± 2% of total quantified PAHs were removed in the weekly- and monthly-fed bioreactors, respectively. Except for BgP, the concentration of each PAH was significantly lower in effluent slurry from the weekly-fed bioreactor than from the monthly-fed bioreactor (P < 0.01). The weekly-fed bioreactor removed 85 ± 1% of the 2- and 3-ring PAHs, 78 ± 2% of the 4-ring PAHs, and 41 ± 3% of the 5- and 6-ring PAHs, while the monthly-fed bioreactor removed 81 ± 2, 64 ± 3, and 19 ± 6% of these groups, respectively.

PAH concentrations in feed soils (black bars), slurry from the monthly-fed bioreactor (grey bars) and slurry from weekly-fed bioreactor (white bars). Values for feed soil are from samples at days 0 and 56, and values for the slurries are from samples at days 28, 56, and 84. Measurements are pooled averages and standard deviations of triplicate soil aliquots at each time point. NAP Naphthalene, ACE acenaphthene, FLU fluorene, PHN phenanthrene, ANT anthracene, FLA fluoranthene, PYR pyrene, BaA benz[a]anthracene, CHR chrysene, BbF benzo[b]fluoranthene, BkF benzo[k]fluoranthene, BaP benzo[a]pyrene, DBA dibenz[a,h]anthracene, BgP benzo[g,h,i]perylene
Analyses of bacterial communities
DNA extracted from effluent slurry and from untreated soil samples provided a means to examine total bacterial communities. DGGE was used first to examine the bacterial communities in each bioreactor as well as the feed soil (Supplementary Information Fig. S1). Examination of the DGGE profiles led to several observations: (a) at day 0 (the start of semi-continuous operation after 50 days of batch treatment), the community profiles were highly similar in each bioreactor and noticeably different from that of the untreated soil; (b) a number of new, prominent bands appeared in the bioreactors subsequent to the introduction of semi-continuous feeding; (c) some bands appeared specific to each bioreactor, suggesting the establishment of different communities; and (d) the weekly-fed bioreactor displayed an overall more consistent community profile than did the monthly-fed bioreactor.
These observations were further investigated by barcoded 16S rRNA gene pyrosequencing, which was used to analyze the bacterial communities present in the feed soil at the beginning and end of semi-continuous operation (days 0 and 140), as well as in the bioreactors at day 0 and each of the monthly sampling events (6 total samples per bioreactor). A total of 118,977 partial gene sequences of sufficient quality and read length were obtained for these 14 libraries. The smallest library (from the monthly-fed bioreactor at day 0) was 4,514 sequences, while the largest (from the day 28 sample of the weekly-fed bioreactor) contained 12,010 sequences.
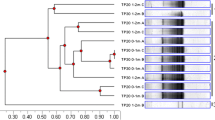
The pyrosequencing data were consistent with the patterns in the DGGE profiles of the samples. Specifically, day 0 samples for both bioreactors contained highly similar types and relative abundances of sequences; both bioreactor communities were significantly different from that in the feed soils; the weekly-fed bioreactor contained a more stable population; and the communities of the weekly-fed and monthly-fed bioreactors, while similar in many respects, also had quantifiable differences. Cluster analysis of the libraries using Fast UniFrac software confirmed and helped visualize these relationships (Fig. 4). The complete table of classified sequences can be found in Supplementary Information (Table S1).

Weighted Fast UniFrac cluster tree showing the relationship of communities in the feed soils and bioreactor samples to one another based on pyrosequence libraries. Abbreviations are the source of the library followed by the sampling time (days after start of semi-continuous operation) in parentheses. Mo Monthly-fed bioreactor, Wk weekly-fed bioreactor, Fd feed soil. The scale bar is the distance between clusters in UniFrac units
The dominant bacterial degraders of six PAHs ranging from two to four rings in the untreated soil used in this study were previously determined by SIP (Jones 2010). These bacterial groups, their PAH growth substrates (as determined by SIP), and their relative abundances in the pyrosequence libraries are presented in Table 1. As might be expected, bacterial groups responded differently to bioreactor conditions. Several phylogenetic groups containing known PAH-degraders were highly represented in the feed soil, but decreased dramatically upon introduction to the bioreactors. The most notable of these were sequences from the genus Pseudoxanthomonas, which comprised 28–33% of the feed soil libraries but represented a maximum of only 1% of the genes in the bioreactors. Other groups exhibiting a similar tendency included the Sphingobium and Pseudomonas genera, as well as members of the uncharacterized Anthracene Group 1 (AG1). Genes from AG1 were the most commonly encountered sequences from 13C-enriched DNA derived from labeled anthracene during DNA-SIP of the untreated Salisbury soil (Jones 2010) and represent uncultivated organisms within the order Sphingomonadales of the Alphaproteobacteria, with closest relation to isolates from the genus Altererythrobacter (~96% 16S rRNA gene similarity).
Conversely, genes from the Sphingomonas genus and the uncultivated Pyrene Group 2 (PG2) increased in relative abundance in the bioreactors compared to the untreated soil. PG2 sequences were particularly abundant in the weekly-fed bioreactor (6–9% of total 16S rRNA genes) compared to the monthly-fed bioreactor (1–4%). Sequences from this uncultivated group, which are closely related Gammaproteobacterial sequences with low similarity to any described genera (~90% 16S rRNA gene similarity to characterized isolates), have been linked with benz[a]anthracene, fluoranthene, and pyrene degradation in this soil by SIP (Jones 2010). PG2 sequences have also been associated with the degradation of pyrene and phenanthrene in other soils (Singleton et al. 2006, 2007; Jones et al. 2008). Genes associated with the Acidovorax genus, which were associated with both naphthalene and phenanthrene degradation in this soil, were highly abundant in both the feed soil and in each of the bioreactors, particularly after the beginning of semi-continuous operation. Overall, bacterial genera or groups containing known PAH-degrading organisms as determined by SIP represented more than 60% of the sequences in the feed soil and from 9 to 37% of the sequences in the bioreactor communities (Table 1).
Other bacterial phylogenetic groups which have thus far not been linked to PAH degradation in this soil but which represented at least 1% of the sequences in at least one pyrosequencing library are listed in Table 2. Some of these groups responded strongly to batch incubation within the bioreactors but later decreased in relative abundance. For example, gene sequences associated with the genus Terrimonas were abundant (7% of all genes) in both bioreactors after the equilibration phase (day 0), but decreased upon the introduction of semi-continuous feeding. A similar phenomenon was observed for unclassified members of the Bradyrhizobiaceae.
After the introduction of the semi-continuous feeding regimes, genes linked to Thiobacillus organisms increased in relative abundance in both bioreactors and remained high throughout the experiment. Genes associated with the Acidobacteria (Group 4 in particular) increased at each time point in the weekly-fed bioreactor (up to 25% relative abundance at the final sampling point) and were generally much more abundant in that community than in the monthly-fed bioreactor community. However, it should be noted that prior to the change in oxygen concentration at day 112, sequences from this group were steadily increasing in relative abundance in the monthly-fed bioreactor as well. Sequences associated with the genus Brevundimonas also comprised a higher percentage of the weekly-fed bioreactor community (5–12% of total genes) compared to the monthly-fed bioreactor (0.4–2%).
Members of the genus Parvibaculum and unclassified members of the Bacteroidetes were the only sequences that were a consistently higher percentage of the monthly-fed bioreactor community than the weekly-fed bioreactor community, particularly in the samples prior to day 112. The majority of these Bacteroidetes sequences possessed high sequence similarity (96%) to uncultivated environmental sequences but only 84% similarity to a named Alistipes species.
The anoxic conditions presumed to temporarily exist in the monthly-fed bioreactor during the day 112 feeding led to a marked shift in the bacterial community, with a dramatic increase in sequences associated with unclassified members of the Gammaproteobacteria. Most of the sequences in this unclassified group (1,702 out of the 6,010 total sequences in the day 112 monthly-fed bioreactor library) were a single clade (at 97% similarity) with <92% sequence similarity to any 16S rRNA gene sequence in the public databases. The closest named relatives to this group were members of the Moraxella and Beggiatoa genera, at 86 and 85% sequence similarity, respectively. There was also a marked decrease in abundance of the unclassified Bacteroidetes sequences in the monthly-fed bioreactor after day 112. During this same time, sequences associated with the genus Sediminibacterium increased to over 4% of the 16S rRNA genes in the monthly-fed bioreactor on day 112 and to over 12% on day 140.
Similar to what had been previously observed in other studies in which contaminated soils were stored for long periods of time (Rost et al. 2002), total PAH concentrations in the feed soil analyzed at day 140 were approximately 18% lower than the average concentrations of days 0 and 56 (data not shown). The most significant decreases were observed for the two- and three-ring PAHs acenaphthene, phenanthrene, and fluorene (32, 28, and 46%, respectively). While these losses might be attributed at least partially to microbial activity, cluster analysis of the pyrosequence libraries confirmed the similarities of the bacterial communities in the feed soils at days 0 and 140 (Fig. 4) and an examination of the groups present in the respective pyrosequence libraries indicated few substantial changes in the feed soil community over the course of the experiment (Tables 1, 2).
Discussion
Semi-continuous aerobic biological treatment of weathered, PAH-contaminated soil removed a high percentage of PAHs present in the initial soil, regardless of the feeding pattern employed. As has been observed by others (Lundstedt et al. 2003; Ringelberg et al. 2001), removal of the LMW PAHs was greater than the removal of the HMW PAHs in both bioreactors. However, the weekly-fed bioreactor removed a higher percentage of the 4-, 5-, and 6-ring PAHs compared to the monthly-fed bioreactor.
In most previous work in which PAH removal from field-contaminated soil has been evaluated in slurry-phase bioreactors, the reactors received a single charge of contaminated soil and were operated in batch mode (Lundstedt et al. 2003; Ringelberg et al. 2001; Rutherford et al. 1998; Talley et al. 2002; Vińas et al. 2005). In one study (Otte et al. 1994), a fed-batch system was used in which contaminated soil was added continuously to an active bioreactor but from which no material was removed. In contrast to strictly batch treatment, fed-batch and semi-continuous bioreactors have the advantage of containing microbial communities selected for the degradation of the primary contaminants in the soil under optimum conditions (mixing and aeration), which is comparable to bioaugmentation with contaminant degraders as a means of increasing the rates of contaminant biodegradation (Otte et al. 1994; Rutherford et al. 1998).
There is limited information in the literature on how the operating characteristics of semi-continuous, slurry-phase bioreactors for soil treatment affect their performance. The work of Cassidy and colleagues (Cassidy et al. 2000, 2002; Cassidy and Hudak 2002) is most relevant to the present study. They demonstrated that semi-continuous reactors were superior to continuous-flow reactors with respect to contaminant removal from soil contaminated with diesel fuel (Cassidy et al. 2000) and PAHs (Cassidy and Hudak 2002). In both cases, the semi-continuous reactor selected for biosurfactant-producing organisms and the associated accumulation of biosurfactants, whereas the continuous-flow reactor did not. They also evaluated the effects of feeding pattern at a fixed residence time on the removal of diesel fuel in semi-continuous, slurry-phase bioreactors (Cassidy et al. 2002). Contaminant removal in their systems was greatest at the highest instantaneous loading of soil in a feeding cycle (50% replacement of the slurry at each feeding). In their systems, however, increased removal of diesel fuel corresponded to increasing production of biosurfactant and the selection of biosurfactant-producing microorganisms (Cassidy et al. 2002). We observed the opposite effect of instantaneous soil loading in the present study, with significantly greater PAH removal at the lower instantaneous loading (weekly feeding with 20% replacement of the slurry).
There are multiple factors which could have contributed to the overall lower performance of the monthly-fed bioreactor. The feeding regime consisted of the removal and replacement of 80% of the slurry during each feeding event. This resulted in the influx of more nitrogen into the system per feeding event than in the weekly-fed bioreactor, a phenomenon that we documented in batch incubations likely contributed to the lower pH of that bioreactor (Fig. 2). The more acidic conditions might have led to reduced activity of PAH-degrading bacteria or selected against their growth. The replacement of such a large volume of slurry also removed the majority of the microbial community that developed during the previous incubation period, creating the potential for greater changes in the community between feeding events than in the weekly-fed bioreactor. The weekly-fed bioreactor maintained a neutral pH and a more consistent bacterial community, and also removed PAHs from the soil to a greater extent than the monthly-fed bioreactor. Overall, however, it is not possible to determine the effects of pH on PAH removal from the available data.
Knowledge of which PAH-degrading bacteria will thrive within the confines of a particular bioremediation scheme can be important in predicting the success of the operation. However, limited work has been done to evaluate PAH-degrading microbial communities in engineered remediation systems. Ringelberg et al. (2001) used phospholipid fatty acid (PLFA) analysis to follow shifts in the microbial community in a batch slurry system treating PAH-contaminated harbor sediment. Their PLFA data suggested an increase in Gram-positive Rhodococcus spp. during the incubation; in our study the Actinobacteria were only minor members of the communities in the feed soil and in the bioreactors. Vińas et al. (2005) used DGGE to observe a substantial shift in the microbial community during batch incubation of a creosote-contaminated soil. Bands cut from the DGGE gels and sequenced were related to members of the genera Xanthomonas, Sphingomonas, Alcaligenes, and Achromobacter, as well as the Bacteroidetes phylum. Of these, only Sphingomonas and various members of the phylum Bacteroidetes were present at levels greater than 1% of the 16S rRNA genes in the bioreactor communities in our study.
To our knowledge, this is the first study to employ barcoded pyrosequencing to evaluate the microbial community in PAH-contaminated soil. Some bacterial groups known to be involved in PAH degradation in this soil were well-represented in the untreated feed soil but were present at lower relative abundance in the bioreactor communities. The most abundant of these sequences were associated with the Pseudoxanthomonas genus. Although Pseudoxanthomonas sequences were previously detected during SIP with naphthalene and anthracene, based on their abundance in clone libraries generated from 13C-enriched DNA during the SIP experiments, members of this genus were not the dominant degraders of either PAH (Jones 2010). Additionally, neither of these PAHs was particularly abundant in the feed soil (Fig. 3) and for naphthalene very little degradation occurred during treatment. This phenomenon of limited naphthalene removal has been observed in previous experiments in our group (Richardson 2010; Zhu and Aitken 2010). We believe that much of the naphthalene in the soil was lost by dissolution in the field environment prior to soil collection and volatilization during soil processing, and that the fraction that remained was not readily bioavailable (Richardson 2010). Other genera containing known PAH-degraders, such as Pseudomonas and Sphingobium, also declined in the bioreactor relative to the untreated soil; therefore, it is unlikely that they were primarily responsible for the bulk of PAH-degradation under bioreactor conditions.
Only three of the known PAH-degrading groups were consistently present in high relative abundance (>1% of total 16S rRNA genes) in both bioreactors during semi-continuous operation: Acidovorax spp., PG2, and Sphingomonas spp. (in order of decreasing representation). The high relative abundance of Acidovorax sequences, which were the dominant sequences identified by SIP with phenanthrene (Jones 2010), in both the feed soil and bioreactor communities was unsurprising given the high concentration of phenanthrene in the soil. Members of the uncultivated PG2, which are now known to degrade pyrene, benz[a]anthracene, fluoranthene, and phenanthrene (Jones et al. 2008; Singleton et al. 2006, 2007; Jones 2010), were present in both bioreactors and may have contributed to the degradation of other PAHs as well. Sphingomonas strains (1–5% of the bioreactor 16S rRNA genes) are well known for their ability to degrade a wide range of PAHs (Stolz 2009), but were associated only with fluoranthene degradation in SIP experiments with this particular soil (Jones 2010). The greatest difference between the two bioreactors among these three groups was in the higher abundance of genes from PG2 in the weekly-fed bioreactor. We hypothesize that this potentially widespread group of undescribed PAH-degraders could be the organisms primarily responsible for greater removal of HMW PAHs in the weekly-fed bioreactor than in the monthly-fed bioreactor.
Organisms from other bacterial groups in the bioreactors may have played a role in PAH degradation even though they were not previously identified as PAH degraders by SIP. The SIP experiments that identified the dominant PAH-degraders in this soil were performed with feed soil that had not been exposed to long-term enrichment, such as the conditions inside a bioreactor, and therefore may have overlooked PAH-degrading organisms that were not abundant in the untreated soil. Some genera that increased substantially in relative abundance during bioreactor operation were Thiobacillus, Parvibaculum, and Brevundimonas. Thiobacillus isolates have been shown to grow in response to phenanthrene amendment of soil (Bodour et al. 2003) and an isolate 96% related to a Parvibaculum strain was recently isolated on pyrene as a sole carbon and energy source (Hilyard et al. 2008). A Brevundimonas strain was also recently shown to grow on phenanthrene (Xiao et al. 2010) and sequences associated with this genus have been observed in several surveys of microbial communities in PAH-contaminated soil or sediment (Chang et al. 2007; Li et al. 2009; Phillips et al. 2008; Vińas et al. 2005). Sequences from each of these genera were well-represented in at least one of the bioreactors but not in the feed soils.
As PAHs make up less than a quarter of the total organic carbon in MGP soil (Haeseler et al. 1999), not all stimulated bacteria in the bioreactors were expected to be associated with PAH degradation. For example, to the best of our knowledge members of the Acidobacteria have not been directly linked to PAH metabolism, so their increasing presence in the bioreactors was likely attributable to growth on other carbon sources. Likewise, sequences related to Sediminibacterium reached over 12% of the total 16S rRNA genes in the monthly-fed bioreactor community by the last sampling event but the sole characterized member of the genus was not reported to grow on PAHs (Qu and Yuan 2008).
References
Agency for Toxic Substances and Disease Registry (2007) 2007 CERCLA priority list of hazardous substances. Agency for Toxic Substances and Disease Registry, Atlanta. http://www.atsdr.cdc.gov/cercla/. Accessed 22 Sep 2010
Bodour AA, Wang JM, Brusseau ML, Maier RM (2003) Temporal change in culturable phenanthrene degraders in response to long-term exposure to phenanthrene in a soil column system. Environ Microbiol 5:888–895
Cassidy D, Hudak A (2002) Microorganism selection and performance in bioslurry reactors treating PAH-contaminated soil. Environ Technol 23:1033–1042
Cassidy D, Efendiev S, White D (2000) A comparison of CSTR and SBR bioslurry reactor performance. Water Res 34:4333–4342
Cassidy DP, Hudak AJ, Murad AA (2002) Effect of loading in soil slurry-sequencing batch reactors on biosurfactant production and foaming. J Environ Eng (ASCE) 128:575–582
Cerniglia CE (1992) Biodegradation of polycyclic aromatic hydrocarbons. Biodegradation 3:351–368
Chang Y-T, Lee J-F, Chao H-P (2007) Variability of communities and physiological characteristics between free-living bacteria and attached bacteria during the PAH biodegradation in a soil/water system. Eur J Soil Biol 43:283–296
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM (2009) The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37:D141–D145
Dean-Ross D, Cerniglia CE (1996) Degradation of pyrene by Mycobacterium flavescens. Appl Microbiol Biotechnol 46:307–312
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Haeseler F, Blanchet D, Durelle V, Werner P, Vandecasteele J-P (1999) Analytical characterization of contaminated soils from former manufactured gas plants. Environ Sci Technol 33:825–830
Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R (2008) Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods 5:235–237
Hamady M, Lozupone C, Knight R (2010) Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J 4:17–27
Hilyard EJ, Jones-Meehan JM, Spargo BJ, Hill RT (2008) Enrichment, isolation, and phylogenetic identification of polycyclic aromatic hydrocarbon-degrading bacteria from Elizabeth River sediments. Appl Environ Microbiol 74:1176–1182
Jones MD (2010) Stable-isotope probing-based investigations of polycyclic aromatic hydrocarbon-degrading bacteria in contaminated soil. PhD dissertation, University of North Carolina at Chapel Hill
Jones MD, Singleton DR, Carstensen DP, Powell SN, Swanson JS, Pfaender FK, Aitken MD (2008) Effect of incubation conditions on the enrichment of pyrene-degrading bacteria identified by stable-isotope probing in an aged, PAH-contaminated soil. Microb Ecol 56:341–349
Kanaly RA, Harayama S (2000) Biodegradation of high-molecular-weight polycyclic aromatic hydrocarbons by bacteria. J Bacteriol 182:2059–2067
Khan AA, Kim SJ, Paine DD, Cerniglia CE (2002) Classification of a polycyclic aromatic hydrocarbon-metabolizing bacterium, Mycobacterium sp. strain PYR-1, as Mycobacterium vanbaalenii sp nov. IJSEM 52:1997–2002
Li H, Zhang Y, Li D, Xu H, Chen G, Zhang C (2009) Comparisons of different hypervariable regions of rrs genes for fingerprinting of microbial communities in paddy soils. Soil Biol Biochem 41:954–968
Lukasewycz MT, Burkhard LP (2005) Complete elimination of carbonates: a critical step in the accurate measurement of organic and black carbon in sediments. Environ Toxicol Chem 24:2218–2221
Lundstedt S, Haglund P, Oberg L (2003) Degradation and formation of polycyclic aromatic compounds during bioslurry treatment of an aged gasworks soil. Environ Toxicol Chem 22:1413–1420
Otte M-P, Gagnon J, Comeau Y, Matte N, Greer CW, Samson R (1994) Activation of an indigenous microbial consortium for bioaugmentation of pentachlorophenol/creosote contaminated soils. Appl Microbiol Biotechnol 40:926–932
Phillips LA, Germida JJ, Farrell RE, Greer CW (2008) Hydrocarbon degradation potential and activity of endophytic bacteria associated with prairie plants. Soil Biol Biochem 40:3054–3064
Price MN, Dehal PS, Arkin AP (2009) FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26:1641–1650
Qu JH, Yuan HL (2008) Sediminibacterium salmoneum gen. nov., sp. nov., a member of the phylum Bacteroidetes isolated from sediment of a eutrophic reservoir. Int J Syst Evol Microbiol 58:2191–2194
Richardson SD (2010) Effects of in situ bioremediation strategies on the biodegradation and bioavailability of polycyclic aromatic hydrocarbons in weathered manufactured gas plant soil. PhD dissertation, University of North Carolina at Chapel Hill
Ringelberg DB, Talley JW, Perkins EJ, Tucker SG, Luthy RG, Bouwer EJ, Fredrickson HL (2001) Succession of phenotypic, genotypic, and metabolic community characteristics during in vitro bioslurry treatment of polycyclic aromatic hydrocarbon-contaminated sediments. Appl Environ Microbiol 67:1542–1550
Rost H, Loibner AP, Hasinger M, Braun R, Szolar OHJ (2002) Behavior of PAHs during cold storage of historically contaminated soil samples. Chemosphere 49:1239–1246
Rutherford PM, Banerjee DK, Luther SM, Gray MR, Dudas MJ, McGill WB, Pickard MA, Salloum MJ (1998) Slurry-phase bioremediation of creosote and petroleum-contaminated soils. Environ Technol 19:683–696
Shumway M, Cochrane G, Sugawara H (2010) Archiving next generation sequencing data. Nucleic Acids Res 38:D870–D871
Singleton DR, Powell SN, Sangaiah R, Gold A, Ball LM, Aitken MD (2005) Stable-isotope probing of bacteria capable of degrading salicylate, naphthalene, or phenanthrene in a bioreactor treating contaminated soil. Appl Environ Microbiol 71:1202–1209
Singleton DR, Sangaiah R, Gold A, Ball LM, Aitken MD (2006) Identification and quantification of uncultivated Proteobacteria associated with pyrene degradation in a bioreactor treating PAH-contaminated soil. Environ Microbiol 10:1736–1745
Singleton DR, Hunt M, Powell SN, Frontera-Suau R, Aitken MD (2007) Stable-isotope probing with multiple growth substrates to determine substrate specificity of uncultivated bacteria. J Microbiol Methods 69:180–187
Singleton DR, Richardson SD, Aitken MD (2008) Effects of enrichment with phthalate on polycyclic aromatic hydrocarbon biodegradation in contaminated soil. Biodegradation 19:577–587
Stolz A (2009) Molecular characteristics of xenobiotic-degrading sphingomonads. Appl Microbiol Biotechnol 81:793–811
Talavera G, Castresana J (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56:564–577
Talley JW, Ghosh U, Tucker SG, Furey JS, Luthy RG (2002) Particle-scale understanding of the bioavailability of PAHs in sediment. Environ Sci Technol 36:477–483
U.S. Environmental Protection Agency (1993) Provisional guidance for quantitative risk assessment of polycyclic aromatic hydrocarbons. Environmental Criteria and Assessment Office, Office of Health and Environmental Assessment, Washington, DC
U.S. Environmental Protection Agency (2004) Cleaning up the nation’s waste sites: markets and technology trends, 2004 edition. U.S. Environmental Protection Agency, Washington, DC
U.S. Environmental Protection Agency (2007) Treatment technologies for site cleanup: annual status report. EPA Office of Superfund Remediation and Technology Innovation, Washington, DC
Vińas M, Sabaté J, Espuny MJ, Solanas AM (2005) Bacterial community dynamics and polycyclic aromatic hydrocarbon degradation during bioremediation of heavily creosote-contaminated soil. Appl Environ Microbiol 71:7008–7018
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Xiao J, Guo L, Wang S, Lu Y (2010) Comparative impact of cadmium on two phenanthrene-degrading bacteria isolated from cadmium and phenanthrene co-contaminated soil in China. J Hazard Mater 174:818–823
Zhu H, Aitken MD (2010) Surfactant-enhanced desorption and biodegradation of polycyclic aromatic hydrocarbons in contaminated soil. Environ Sci Technol 44:7260–7265
Zhu H, Roper JC, Pfaender FK, Aitken MD (2008) Effects of anaerobic incubation on the desorption of polycyclic aromatic hydrocarbons from contaminated soils. Environ Toxicol Chem 27:837–844
Acknowledgment
This work was supported by the U.S. National Institute of Environmental Health Sciences (NIEHS; grant number 5 P42 ES005948).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Singleton, D.R., Richardson, S.D. & Aitken, M.D. Pyrosequence analysis of bacterial communities in aerobic bioreactors treating polycyclic aromatic hydrocarbon-contaminated soil. Biodegradation 22, 1061–1073 (2011). https://doi.org/10.1007/s10532-011-9463-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10532-011-9463-3