
Abstract
The goldspotted oak borer, Agrilus auroguttatus Schaeffer, is an invasive woodborer in California USA that is native to oak woodlands across southern Arizona USA. Developing a classical biological control program for this pest in southern California is a high priority due to the continuing ecological and economic damage caused by this insect since its recent introduction into the area. In an attempt to determine the area of origin for this invasive beetle, analyses of the mitochondrial cytochrome oxidase and ribosomal nuclear D2 domain of the 28S gene regions were undertaken and provided insight into the phylogeographic relationship between and within populations of A. auroguttatus in Arizona and California. The area of origin for the invasive population of goldspotted oak borer in California was not determined conclusively, although our molecular data suggests the Dragoon Mountains in Cochise Co., Arizona as a possible source for the California population of A. auroguttatus. Results also confirmed that individuals collected from populations across southern Arizona and California are all A. auroguttatus, and are not part of a cryptic species complex comprised of the morphologically similar A. coxalis. Future surveys for natural enemies of A. auroguttatus will focus on the Dragoon Mountains as a potential source for co-evolved enemies for use in a classical biological control program against this invasive woodborer in southern California.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The goldspotted oak borer, Agrilus auroguttatus Schaeffer, (Coleoptera: Buprestidae) is an invasive wood-boring beetle that aggressively attacks native oak trees in southern California, USA. Native to Arizona, this beetle was initially detected in the Descanso Ranger District, Cleveland National Forest (DRD-CNF), San Diego County, California, in 2004, but was likely introduced accidentally several years earlier through movement of infested oak firewood (Coleman and Seybold 2008a; Coleman et al. 2012a). Infestation of A. auroguttatus in southern California currently covers approximately 213,000 ha across San Diego, and Riverside Counties (Jones et al. 2013), and mortality to native coast live oak, (Quercus agrifolia Nee), California black oak (Q. kelloggii Newb.), and canyon live oak (Q. chrysolepis Liebm) from A. auroguttatus, caused by extensive larval feeding damage to the phloem/xylem interface, is estimated at 25,000 trees (Coleman et al. 2012b; USDA Forest Health Monitoring [FHM] 2013). The documentation of oak mortality since 2002 indicates that the distribution of this beetle is radially expanding within San Diego Co. (USDA FHM 2013), which can mostly be attributed to natural dispersal. However, satellite infestations in Marion Bear Memorial Park, San Diego Co. (approximately 32 km from the closest known infestation) in 2009, and in Idyllwild, Riverside Co. (approximately 70 km from the closest known infestation) in 2012 are hypothesized to have resulted from further movement of infested oak firewood (Jones et al. 2013), indicating that this is a viable long-distance dispersal mechanism for A. auroguttatus in California (Lopez et al. 2014).
The introduction and establishment of invasive wood-boring and bark beetles has become increasingly problematic in recent decades (Liebhold et al. 1995; Perrings et al. 2005; Brockerhoff et al. 2006; Hulme et al. 2008; Tobin et al. 2010). Invasive pests such as the emerald ash borer, Agrilus planipennis Fairmaire, Asian longhorn beetle, Anoplophora glabripennis (Motschulsky), red bay ambrosia beetle, Xyleborus glabratus Eichhoff, and the red turpentine beetle, Dendroctonus valens LeConte, have caused a tremendous amount of ecological and economical damage after being introduced in non-native habitats through the transportation of infested wood material (Yan et al. 2005; Poland and McCullough 2006; Haack et al. 2010; Hulcr and Dunn 2011). Managing these invasive species in natural ecosystems is challenging because at the landscape level, the majority of tools available (i.e., chemical or physical/mechanical control) work only if the infested area is small or sufficiently isolated to prevent re-infestation (Van Driesche et al. 2010). Eradication of newly introduced, isolated populations of invasive wood-boring and bark beetles in a forest environment is typically not successful because of the difficulty in detecting these invaders at an early stage. Concealed inside trees or other wood material, these insects are often overlooked until extensive decline and mortality occurs within tree hosts (Coleman and Seybold 2008a; Kovacs et al. 2010). Classical biological control can be an efficient management strategy for controlling invasive species in natural ecosystems since biological control agents have the ability to become established and remain in the environment indefinitely, providing long-term widespread management of the target pest without continuous human intervention (Van Driesche et al. 2010).
Molecular markers have the potential to supply valuable information for making management decisions about invasive species by characterizing population level genetic variation, which in turn can aid reconstruction of the history of an invasion, and identify the geographic origin of the invading population (Cognato et al. 2005; Rugman-Jones et al. 2007; Simonsen et al. 2008; Bray et al. 2011; Rugman-Jones et al. 2012). This information can be especially useful when developing a classical biological control program because, in some instances, little is known about a species prior to its establishment and subsequent spread in a non-native habitat. Insight into the invasion history and source of an invasive pest can help to identify transport vectors and direct the search for co-evolved biological control agents (Rugman-Jones et al. 2007). Understanding genetic variation within and among populations of an invasive species (in both the native and introduced range) can also help clarify taxonomic identity of a pest. For example, molecular analyses reported in Coleman et al. (2012a) helped resolve long-running taxonomic confusion between A. auroguttatus and its morphologically similar congener A. coxalis Waterhouse (Hespenheide et al. 2011), and narrowed the native distribution of A. auroguttatus to southern Arizona, and perhaps northern Mexico (based on a single specimen collected from Baja California Sur in 1977) (Hespenheide et al. 2011), though this suspected Baja population has yet to be confirmed (Coleman et al. 2012b).
Within southern Arizona, records indicate that A. auroguttatus has been collected from several mountain ranges including the Chiricahua, Huachuca, Santa Catalina, and Santa Rita mountains since the early 1900s (Coleman and Seybold 2011). These mountain ranges can be considered “sky islands”, isolated by the surrounding Sonoran Desert, which likely presents a dispersal barrier for A. auroguttatus and its natural enemies. The first collection of A. auroguttatus from the Dragoon mountains in 2011 suggested there are additional mountain ranges within the greater home range that may be suitable for A. auroguttatus inhabitation, and some of these (e.g., the Whetstone and Patagonia Mountains) have not been systematically surveyed for this beetle. Narrowing the geographic source of the invasive California population to a specific mountain range in Arizona would, in theory, allow the collection of natural enemies which have formed a co-evolutionary relationship with the genotype of the invasive population (Stouthamer 2008).
The mitochondrial cytochrome oxidase (COI) gene has been useful in studies investigating the origin of an invasive population (Havill et al. 2006; Rugman-Jones et al. 2007; Cognato et al. 2005). With corroborative support from other independently evolving gene regions (e.g., the highly conserved nuclear ribosomal 28S gene), COI has also been used to separate species which are morphologically indistinguishable (Rugman-Jones et al. 2010; Coleman et al. 2012a). In the present study, we utilized sequences of COI and the D2 domain of the 28S gene (28SD2) to investigate connectivity among A. auroguttatus populations from the San Jacinto Mountains and Cleveland National Forest in southern California, and the Chiricahua, Dragoon, Huachuca, Santa Catalina, and Santa Rita Mountains in southern Arizona. Our goal was to better understand genetic variation within and between populations in the native and introduced ranges, with the hope of identifying the geographic origin of the invasive California population. Determining the area of origin for A. auroguttatus is a fundamental step in the search for co-evolved natural enemies of this invasive pest that may be considered for use in a classical biological control program in southern California.
Materials and methods
Specimen collections
Specimens of A. auroguttatus were collected between May 2009 and November 2012 from infested oak trees in San Diego and Riverside Counties, California, and across several mountain ranges in southern Arizona, USA. (Table 1; Fig. 1). In California, adult specimens were either recovered from purple and green prism sticky traps placed throughout the DRD-CNF, San Diego Co., or reared from infested Q. agrifolia and Q. kelloggii material collected from this area (Coleman and Seybold 2011). In addition, all life stages of A. auroguttatus were collected by destructively sampling infested Q. agrifolia and Q. kelloggii trees found in DRD-CNF, and in the town of Idyllwild, in the San Jacinto Mountains, Riverside Co. Arizona specimens were also collected by destructive sampling or reared from infested Q. emoryi and Q. hypoleucoides (Coleman et al. 2012a). All collected specimens (live and dead) were placed into 95 % ethanol and stored at −20 °C to preserve DNA for analysis. Voucher specimens were deposited in the University of California, Riverside Entomology Museum (UCRC ENT 407267–UCRC ENT 407284).

Map of collection sites for A. auroguttatus in southern California and Arizona. See Table 1 for map legend with corresponding collection information
DNA extractions
Whole genomic DNA was extracted from individual specimens using a standard Chelex method (Walsh et al. 1991). Individual A. auroguttatus were removed from ethanol and allowed to air-dry on filter paper. A tissue sample was then taken from each specimen and transferred to a 0.5 μl microcentrifuge tube containing 4 μl of proteinase-K. Tissue samples comprised either a single hind tibia for adult specimens, or a small slice (approx. 1–3 mm3) from the head capsule of larval specimens. Tissue samples were homogenized in the proteinase-K using a micropestle, after which, 100 μl of a 5 % Chelex® 100 (Bio-Rad Laboratories, Hercules, CA) suspension (in water) was added and the tubes were incubated at 55 °C for 1 h, then at 99 °C for an additional 10 min to inactivate the proteinase-K. The chelex resin and insect debris were pelleted in a microcentrifuge at 14,000 rpm for 4 min, and the supernatant (containing the extracted DNA) was transferred to a new 0.5 μl microcentrifuge tube and stored at −20 °C.
Amplification of extracted A. auroguttatus DNA
Genetic variation across California and Arizona populations of A. auroguttatus was examined by amplifying a section of the mitochondrial gene (mtDNA) COI c subunit 1 using the polymerase chain reaction (PCR). Reactions were performed in 25 μl volumes containing 2 μl of DNA template (concentration not determined), 1 ThermoPol PCR Buffer (New England BioLabs, Ipswich, MA, USA), an additional 1 mM MgCl2, 200 μM each of dATP, dCTP, and dGTP, and 400 μM dUTPl, 4 % (v/v) BSA (NEB), 1 U Taq polymerase (NEB), and 0.2 μM each of the primers LCO1490 (5′-GGTCAACAAATCATAAAGATATTGG-3′) and HCO2198 (5′-TAAACTTCAGGGTGACCAAAAAATCA-3′) (Folmer et al. 1994). PCR was performed in a Mastercycler® ep gradient S thermocycler (Eppendorf North America Inc., New York, NY, USA) with the following settings: 2 min at 94 °C; followed by five cycles of 30 s at 94 °C, 1 min 30 s at 45 °C, and 1 min at 72 °C; followed by a further 35 cycles of 30 s at 94 °C, 1 min 30 s at 51 °C, and 1 min at 72 °C; and a final extension of 5 min at 72 °C. A section of the 28SD2 nuclear ribosomal DNA was also amplified from a subsample of specimens collected from each locality and representing a broad diversity of COI haplotypes (see Results), using the 28sF3663 and 28sR4076 primers and protocol described in Rugman-Jones et al. (2010).
Cleaning and sequencing
Successful amplification was confirmed by electrophoresis on 1 % agarose gels stained with ethidium bromide. PCR products were subsequently cleaned using the Wizard® PCR Preps DNA purification system (Promega, Madison, WI, USA) and sequenced in both directions at the Institute for Integrative Genome Biology, University of California Riverside. Sequences were aligned manually in BioEdit 7.0.5.3 (Hall 1999) and primer sequences were removed prior to analysis. This resulted in an aligned COI matrix of 286 sequences, each 658 bp long, and an aligned 28S matrix of 23 sequences, each 536 bp long. Representative COI and 28S sequences were deposited in GenBank (Benson et al. 2008) (accession numbers: JF719839–JF719885 and KF705201–KF705204).
Genetic analysis
Cytochrome oxidase sequences were translated using the EMBOSS-Transeq website (http://www.ebi.ac.uk/Tools/emboss/transeq/index.html) to confirm the absence of nuclear pseudogenes (Song et al. 2008). A haplotype (H) network was constructed using TCS version 1.21 with default run settings (Clement 2000). Genetic diversity of the entire sequence set, and within and between each population was examined by calculating the average number of nucleotide substitutions per site using Dnasp v. 5.10.1 (Librado and Rozas 2009). Population pairwise estimates of ΦST, based on a matrix of uncorrected pairwise distances, were also calculated in ARLEQUIN v. 3.11. The significance of ΦST was evaluated using the permutation method (10,000 permutations) invoked in the ARLEQUIN program (Bonferrroni adjusted α = 0.0024). ARLEQUIN was also used to perform an analysis of molecular variation (AMOVA) (Excoffier et al. 1992), partitioning total genetic variation into three hierarchical component levels: within populations, among populations, and among native and introduced population groups. Again, significance of the AMOVA was evaluated using 10,000 permutations.
Results
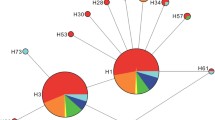
A section of the COI gene of 286 A. auroguttatus individuals (adults and larvae), 147 from Arizona and 139 from California, was sequenced. Among these sequences, a total of 39 haplotypes were identified. Haplotype distribution, abundance, and diversity across sample locations in Arizona and California is shown in Fig. 2 and Table 2. The majority of haplotypes grouped into three distinct genetic clusters (Fig. 2). The first cluster comprised 17 haplotypes centered on the most common haplotype (H1), with all haplotypes no more than 2 bp distant. The second cluster again comprised 17 haplotypes, this time centered around H8, with 12 of the 16 “satellite” haplotypes being no more than 2 bp distant, and the most distant (H14) being 5 bp distant. Haplotypes from clusters one and two were separated by at least six nucleotide substitutions. The third cluster, comprised of three haplotypes (H31, H21 and H19), was detected only in the Chiricahua Mountains, AZ; the easternmost part of the sampled range. Of the remaining two haplotypes, H29 fell between clusters one and two, while H25 was relatively distant from all other haplotypes, at 9 bp from its closest genetic neighbor (H31). Genetic variation across the entire sequence set (average number of nucleotide substitutions per site = 0.0094) was well within the expectations of a single species. Furthermore, all sequences of 28SD2 obtained in this study were identical. This constituted 23 individuals sampled from the Chiricahua (n = 1), Huachuca (n = 1), Santa Catalina (n = 1), and Santa Rita (n = 6) Mountains in Arizona, and from the Cleveland National Forest (n = 9), and San Jacinto (n = 5) Mountains in California. Together, this strongly suggests that individuals collected from these populations were all A. auroguttatus.

Mitochondrial haplotype network for A. Auroguttatus individuals collected in California and Arizona, USA. Each haplotype is represented by a rectangle or oval. Haplotype size is proportional to the number of specimens sharing a haplotype, and the rectangular haplotype is that assigned to the highest outgroup probability. Small circles unobserved inferred haplotypes and lines between haplotypes represent a single nucleotide mutational change. Alpha-numeric codes and their corresponding localities are shown in Table 1
Despite this apparent genetic structure, there was little evidence of any geographic signal in the major clustering of the haplotypes (Fig. 2). Consequently, there was little overlap in the genetic and geographic distributions of individual haplotypes. This was evident in our estimates of ΦST (Table 3) with 18 of 21 pairwise comparisons exceeding 0.18, suggesting very low levels of gene flow between native populations. Indeed, only two haplotypes were shared between CA and AZ, one from each cluster. The most common haplotype (H1) was shared by eighty-two specimens and found in every sample location except the Santa Catalina Mountains, AZ. However, the second overlapping California and Arizona haplotype (H34) was shared only between individuals collected from the Dragoon Mountains in Arizona (n = 1) and the Cleveland National Forest in California (n = 4). The remaining haplotypes were either from specimens collected only in Arizona (n = 23) or only in California (n = 14).
Analysis of molecular variance (Table 4) revealed that the greatest amount of total nucleotide variation (67.49 %) was accounted for by differences among individuals within populations. A smaller but significant amount of variation (24.68 %) was found among populations within groups, but variation between native and introduced groups was not significant.
Discussion
The recent introduction of A. auroguttatus has resulted in the mortality of tens of thousands of mature red oaks (section Lobatae) in southern California, which is drastically changing the composition of these important and unique oak woodland communities (Allen-Diaz et al. 2007; Coleman et al. 2012b). The difficulty in managing this wood-boring pest in a natural ecosystem has underscored the importance of developing a classical biological control program for A. auroguttatus in southern California. Analysis of DNA sequences from the COI and 28SD2 gene regions allowed the comparison of A. auroguttatus populations in California and across several mountain ranges in Arizona in an attempt to identify the source of this invasive beetle. The identical nature of 28SD2 sequences from specimens across the sampled geographic range indicated that individuals collected in California and Arizona were all one species, and not the congener A. coxalis or perhaps another ‘unknown’ cryptic species morphologically indistinguishable from A. auroguttatus or A. coxalis (Coleman et al. 2012a). The identification of 39 haplotypes from 286 individuals across all sample locations showed there was high variability in the COI gene region of this species, but, except for the most abundant haplotype (H1), the distribution of the haplotypes revealed little genetic overlap between the native and introduced regions. Therefore, of the native populations sampled, the Dragoon Mountains in the Coronado National Forest, Arizona, was the only one to share additional haplotypes with the invasive Californian populations, highlighting this population as a likely source of A. auroguttatus in southern California.
The mixture of California and Arizona haplotypes among the three clusters in Fig. 2 provides little evidence of any geographic signal in the major grouping of haplotypes in this network. Surprisingly, 37 of 39 haplotypes sampled across California (n = 14) and Arizona (n = 23) populations were distinct to either region. Out of the 23 distinct haplotypes identified from the Arizona collections, 17 were unique to either the Chiricahua (n = 4), Dragoon (n = 1), Huachuca (n = 7), Santa Catalina (n = 3), or Santa Rita (n = 2) Mountains, supporting the idea that populations of A. auroguttatus from these “sky islands” in southern Arizona are genetically isolated from one another. The lack of gene flow between native populations of A. auroguttatus revealed by our data justifies the use of population genetics to focus the search for co-evolved natural enemies of this woodborer.
In California, 13 unique haplotypes were identified from collections in the Cleveland National Forest in San Diego Co., and a single unique haplotype was collected in the San Jacinto Mountains, Riverside Co., California. The relatively high genetic variation observed in the introduced population of A. auroguttatus is an unexpected result since a substantial decrease in genetic diversity is often the result of founder events (Slade and Moritz 1998; Gwiazdowski et al. 2006; Havill et al. 2006). However, high genetic variation was also observed in the introduced range of the invasive red turpentine beetle, Dendroctonus valens, in China (Cognato et al. 2005). These results suggest that a single, large introduction, and/or multiple small introductions from different regions in southern Arizona could account for the high genetic variation observed in introduced A. auroguttatus populations. Furthermore, within our sample, 31 specimens collected from the bark and phloem of a single tree in Arizona yielded six haplotypes (data not shown). Given this high level of diversity in a single tree in Arizona, it is possible that the California population may have arisen from a single geographic source point, perhaps even from just a single infested tree in Arizona.
Two haplotypes were shared between populations in California and Arizona indicating the presence of some genetic similarity between the native and introduced range. The most common haplotype (H1) was shared between eighty-two specimens, and was detected in every population sampled, except the Santa Catalina Mountains in Arizona. The prevalence of this haplotype throughout the native and introduced range does not help delineate the origin of the invasive California population, since it is likely to occur in most regions within the native range. However, the second overlapping California and Arizona haplotype (H34) was shared only between individuals collected from the Dragoon Mountains in Arizona (n = 1) and the Cleveland National Forest in California (n = 4). Due to minimal overlap between haplotypes identified from California and Arizona, the shared H34 haplotype may indicate that the Dragoon Mountains in Arizona is the most likely source of the invasive California population, and consequently should be an area of focus when surveying for natural enemies of A. auroguttatus. However, it seems likely that we have not yet sampled the full range of variation in this species (see Table 2), and larger samples including collections across additional unsampled mountain ranges could help to further pinpoint the geographic source of the California population of A. auroguttatus. Additionally, we cannot rule out the possibility that the ‘true’ source of the invasive population currently remains unsampled. One possibility is that A. auroguttatus originated from a population in northern Mexico near the Arizona border. Little is known of the distribution of A. auroguttatus in this region, but it is possible that A. auroguttatus may occupy similar “sky islands” in the Sonoran Desert as it stretches south across the border (see Fig. 1). To resolve uncertainty over the area of origin, additional collections from the Chiricahua, Dragoon, Huachuca, Santa Catalina, Patagonia, and Whetstone Mountain ranges in Arizona and from the Sierra San Luis, Sierra El Tigre, and Sierra Madre mountains in northeastern Sonora, Mexico are recommended. It is also feasible that a single variable marker, in this case the maternally inherited COI (analyses using this gene region assume that dispersal of males and females is the same), does not contain enough information to accurately predict the source of the invasive A. auroguttatus populations in California, and analysis of additional markers may be required. The development and use of microsatellites, or the newly developing field of RAD sequencing may offer a better understanding of the genetic variation (Puritz et al. 2012) among A. auroguttatus populations, and help to more conclusively determine the area of origin for this invasive pest.
References
Allen-Diaz BH, Standiford RB, Jackson RD (2007) Oak woodlands and forests. In: Barbour MG, Keeler-Woolf T, Schoenherr AA (eds) Terrestrial vegetation of California. University of California Press, Berkeley, pp 313–338
Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL (2008) GenBank. Nucleic Acids Res 28:15–18
Bray AM, Bauer LS, Poland TM, Haack RA, Cognato AI, Smith JJ (2011) Genetic analysis of emerald ash borer (Agrilus planipennis Fairmaire) populations in Asia and North America. Biol Invasions 13:2869–2887
Brockerhoff EG, Liebhold AM, Jactel H (2006) The ecology of forest insect invasions and advances in their management. Can J For Res 36:263–268
Clement M (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Cognato AI, Sun JH, Anducho-Reyes MA, Owen DR (2005) Genetic variation and origin of the red turpentine beetle (Dendroctonus valens LeConte) introduced to the People’s Republic of China. Agric For Entomol 7:87–94
Coleman TW, Seybold SJ (2008a) New pest in California: the goldspotted oak borer, Agrilus coxalis Waterhouse. USDA Forest Service, Pest Alert, R5-RP-022. 28 Oct 2008, p 4
Coleman TW, Seybold SJ (2011) Collection history and comparison of the interactions of the goldspotted oak borer, Agrilus auroguttatus Schaeffer (Coleoptera: Buprestidae), with host oaks in southern California and southeastern Arizona, USA. Coleopt Bull 65:93–108
Coleman TW, Lopez V, Rugman-Jones P, Stouthamer R, Seybold SJ, Reardon R, Hoddle MS (2012a) Can the destruction of California’s oak woodlands be prevented? Potential for biological control of the goldspotted oak borer, Agrilus auroguttatus. Biocontrol 57:211–225
Coleman TW, Graves AD, Hoddle M, Heath Z, Chen Y, Flint ML, Seybold SJ (2012b) Forest stand impacts associated with Agrilus auroguttatus Schaffer (Coleoptera: Buprestidae) and Agrilus coxalis Waterhouse in oak woodlands. For Ecol Manag 276:104–117
Excoffier L, Smouse P, Quattro J (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3:294–299
Gwiazdowski RA, Van Driesche RG, Desnoyers A, Lyon S, Wu SA, Kamata N, Normark BB (2006) Possible geographic origin of beech scale, Cryptococcus fagisuga (Hemiptera: Eriococcidae), an invasive pest in North America. Biol Control 39:9–18
Haack RA, Hérard F, Sun J, Turgeon JJ (2010) Managing invasive populations of Asian long horned beetle and citrus long horned beetle: a worldwide perspective. Annu Rev Entomol 55:521–546
Hall TA (1999) BioEdit: a user friendly biological sequence alignment and analysis program from Windows 95/98/NT. Nucleic Acids Symp 41:95–98
Havill NP, Montgomery ME, Yu G, Shiyake S, Caccone A (2006) Mitochondrial DNA from hemlock woolly adelgid (Hemiptera: Adelgidae) suggests cryptic speciation and pinpoints the source of the introduction to eastern North America. Ann Entomol Soc Am 99:195–203
Hespenheide HA, Westcott RL, Bellamy CL (2011) Agrilus Curtis (Coleoptera: Buprestidae) of the Baja California peninsula, México. Zootaxa 2805:36–56
Hulcr J, Dunn R (2011) The sudden emergence of pathogenicity in insect-fungus symbioses threatens naive forest ecosystems. Proc R Soc B 278:2866–2873
Hulme PE, Bacher S, Kenis M, Klotz S, Kühn I, Minchin D, Nentwig W, Olenin S, Panov V, Pergl J, Pyšek P, Roques A, Sol D, Solarz W, Vilà M (2008) Grasping at the routes of biological invasions: a framework for integrating pathways into policy. J Appl Ecol 45:403–414
Jones MI, Coleman TW, Graves AD, Flint ML, Seybold SJ (2013) Sanitation options for managing oak wood infested with the invasive goldspotted oak borer (Coleoptera: Buprestidae) in Southern California. J Econ Entomol 106:235–246
Kovacs KF, Haight RG, McCullough DG, Mercader RJ, Siegert NW, Liebhold AM (2010) Cost of potential emerald ash borer damage in US communities, 2009–2019. Ecol Econ 69:569–578
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Liebhold AM, MacDonald WL, Bergdahl D, Mastro VC (1995) Invasion by exotic forest pests: a threat to forest ecosystems. For Sci Monogr 30:1–49
Lopez VM, McClanahan MNL, Graham L, Hoddle MS (2014) Assessing the flight capabilities of the goldspotted oak borer, Agrilus auroguttatus (Coleoptera: Buprestidae), with computerized flight mills. J Econ Entomol (in press)
Perrings C, Dehnen-Schmutz K, Touza J, Williamson M (2005) How to manage biological invasions under globalization. Trends Ecol Evol 20:212–215
Poland TM, McCullough DG (2006) Emerald ash borer: invasion of the urban forest and the threat to North America’s ash resource. J For 104:118–124
Puritz JB, Addison JA, Toonen RJ (2012) Next-generation phylogeography: a targeted approach for multilocus sequencing of non-model organisms. PLoS ONE 7:e34241
Rugman-Jones PF, Hoddle MS, Stouthamer R (2007) Population genetics of Scirtothrips perseae: tracing the origin of a recently introduced exotic pest of Californian avocado orchards, using mitochondrial and microsatellite DNA markers. Entomol Exp Appl 124:101–115
Rugman-Jones PF, Hoddle MS, Stouthamer R (2010) Nuclear mitochondrial barcoding exposes the global pest western flower thrips, Frankliniella occidentalis (Thysanoptera: Thripidae) as two sympatric cryptic species in its native California. J Econ Entomol 103:877–886
Rugman-Jones PF, Hoddle MS, Phillips PA, Jeong G, Stouthamer R (2012) Strong genetic structure among populations of the invasive avocado pest Pseudacysta perseae (Heidemann) (Hemiptera: Tingidae) reveals the source of introduced populations. Biol Invasions 14:1079–1100
Simonsen TJ, Brown RL, Sperling FA (2008) Tracing an invasion: phylogeography of Cactoblastis cactorum (Lepidoptera: Pyralidae) in the United States based on mitochondrial DNA. Ann Entomol Soc Am 101:899–905
Slade RW, Moritz C (1998) Phylogeography of Bufo marinus from its natural and introduced ranges. Proc R Soc Lond Ser B 265:769–777
Song H, Buhay JE, Whiting MF, Crandall KA (2008) Many species in one: DNA bar coding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. Proc Natl Acad Sci 105:13486–13491
Stouthamer R (2008) Molecular tools. In: Van Driesche R, Hoddle M, Center T (eds) Control of pests and weeds by natural enemies: an introduction to biological control. Blackwell, Malden, pp 167–179
Tobin PC, Diss-Torrance A, Blackburn LM, Brown BD (2010) What does “local” firewood buy you? Managing the risk of invasive species introduction. J Econ Entomol 103:1569–1576
USDA Forest Service (FS), Forest Health Monitoring (FHM) (2013) Aerial Survey Region 5 database. (http://www.fs.usda.gov/detail/r5/forest-grasslandhealth/?cid=fsbdev3_046696)
Van Driesche RG, Carruthers RI, Center T, Hoddle MS, Hough-Goldstein J, Morin L, Smith L, Wagner DL et al (2010) Classical biological control for the protection of natural ecosystems. Biol Control Suppl 1:S2–S33
Walsh PS, Metzger DA, Higuchi R (1991) Chelex®100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10:506–513
Yan Z, Sun J, Don O, Zhang Z (2005) The red turpentine beetle, Dendroctonus valens LeConte (Scolytidae): an exotic invasive pest of pine in China. Biodivers Conserv 14:1735–1760
Acknowledgments
We thank Mike Lewis, Allison Bistline, and Ruth Amrich (University of California, Riverside). We are also grateful to Laurel Haavik (Great Lakes Forestry Ctr, Sault Ste Marie, Canada), Andrew Graves (USDA Forest Service-Forest Health Protection, Region 3), and Michael Jones (USDA Forest Service-Forest Health Protection, Region 5) for help with specimen collections. Funding for this study was supported, in part, by two agreements between the USDA Forest Service and the UC Riverside Department of Entomology: Cooperative Agreement # 09-CA-11420004-357 and Joint Venture Agreement # 10-JV-11272172-059. The comments of anonymous reviewers greatly improved earlier versions of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lopez, V.M., Rugman-Jones, P.F., Coleman, T.W. et al. Population genetics of goldspotted oak borer, Agrilus auroguttatus Schaeffer (Coleoptera: Buprestidae): investigating the origin of an invasive pest of native oaks in California. Biol Invasions 16, 2393–2402 (2014). https://doi.org/10.1007/s10530-014-0672-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-014-0672-7