
Abstract
The brown anole, Anolis sagrei, is one of the most widespread and successful colonisers of the diverse Anolis genus, which comprises c. 400 species occurring naturally in Central and South America and the Caribbean. Based on extensive between and within population sampling from a previously published study (334 mitochondrial DNA sequences) and sampling for this study (37 mtDNA sequences), we reconstruct a phylogeny and produce a haplotype network to assign a recently introduced population in St Vincent, Lesser Antilles to its geographic origin. A single haplotype was present in the St Vincent population, which was identical to a haplotype from Tampa, FL. We show that genetic diversity within native range populations, combined with low frequencies of introduced haplotypes in native ranges, may impair attempts to identify source populations, even despite intensive sampling effort. The absence of mtDNA haplotype diversity suggests a significant genetic founder effect within the St Vincent population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As human activity increasingly shapes the distribution and abundance of species at scales ranging from landscape to global, an understanding of the ecological consequences must follow if we are to respond appropriately to conservation issues raised by these biogeographical alterations. The significant and wide-ranging consequences of anthropogenic influences on biogeographic patterns, biodiversity and ecosystem stability are exemplified in human-mediated species introductions. Once established, introduced species can have significant ecological and evolutionary impacts on native species, communities and ecosystems, and severe consequences for the economic welfare of invaded environments (Sakai et al. 2001). In recent years, it has been increasingly recognised that the genetic diversity and demographic stability of introduced populations are key indicators of the potential for self-sustaining establishment in novel environments (Suarez and Tsutsui 2008). Thus, population genetic and demographic investigations are valuable in indicating the requirement and direction for conservation focus in empirical introductions (Costa-Pierce 2003).
The rapid increase in global anthropogenic movements over the past century has led to an associated dramatic rise in the number of reports of species outside their natural ranges (Allendorf and Lundquist 2003). Although widespread commercial transportation has provided copious opportunities for species rafting, proportionately few imported populations successfully establish in new environments (Shigesada and Kawasaki 1997). Past investigations of this phenomenon have focused on the susceptibility of environments and characterisation of invasive species traits to account for this differential establishment ability (Schloesser et al. 1996). Furthermore, it has been recently recognised that imported species are frequently exposed to novel selective pressures in the new environment (Lee 2002). Although genetic characterisations of introduced populations (e.g. using mitochondrial DNA sequencing) are based on neutral genetic variation, they can infer whether a degree of genomic genetic diversity exists, in turn indicating a potential for adaptive evolutionary change (Sakai et al. 2001). The identification of the geographic source for introduced non-natives using phylogenetic techniques is fundamental to elucidating the influence of multiple introductions, gene flow and hybridisation on introduction success in addition to the reconstruction of introduction histories (Durka et al. 2005). Moreover, genetic analyses of introduced founder populations have valuable applications outside the field of biological invasions. Molecular investigations of founder populations also provide an invaluable model for the genetic consequences of demographic bottlenecks faced by many endangered species and fragmented populations (Frankham et al. 2002), and contribute to the understanding of natural colonisation and radiation processes.
Human-mediated invasions have often been documented within decades of initial import (Williams 1969). It is less common, however, to discover an invasion less than 5 years from the initial importation. Reports of the non-native Anolis sagrei (Sauria: Iguanidae) on the island of St Vincent, Lesser Antilles, dating back to 2003 (F. Providence, personnel communication), have provided a fortuitous opportunity for investigating the genetic processes involved in the early stages of introductions. Anoles have been widely transported throughout the Caribbean both by natural (Calsbeek and Smith 2003; Stenson et al. 2004) and human mediated dispersal (Campbell 1996). However, the St Vincent discovery is rare in that the single point of import is known, its current range within the island is small, and two native competitors (A. trinitatis and A. griseus) already occupy and partition Anolis niche space on the island (Schoener and Gorman 1968).
A. sagrei has been known to be a widespread and successful coloniser (Williams 1969). It has spread throughout the entire Florida peninsula (Campbell 2003), well into the south eastern US (see historical overview in Campbell and Echternacht 2003) and into mainland Belize/Mexico (Lee 1992). It has even reached some very isolated islands including Hawaii (McKeown 1996), Taiwan (Norval et al. 2002), Grenada (Greene et al. 2002) and St Vincent (this paper). An extensive phylogeographical study by Kolbe et al. (2004) identified the source of many of these populations using mitochondrial DNA data, and indicated that the high genetic diversity in introduced populations may result from multiple introductions from genetically different native populations.
In this study we use molecular sequencing techniques to reveal the source population for introduced A. sagrei in St Vincent. A phylogeny of A. sagrei was produced from mitochondrial DNA (mtDNA) sequence data comprised of haplotypes obtained from new sampling in St Vincent and published sequences for this species from throughout the native and introduced range (Kolbe et al. 2004). Qualities such as relatively low effective population size and rapid mutation rate make mtDNA useful for assessing genetic relationships of individuals or groups within a species, accounting for the popularity of mtDNA as a marker in phylogeographic studies (Avise 1994). We use haplotype networks and phylogeographic analysis to reveal the likely geographical origin for the introduced population, and discuss the impact of native range within-population genetic diversity on the reliability of identifying geographical source populations. We compare the genetic diversity of the St Vincent population to other (including A. sagrei) introductions, suggest its basis, and discuss the relative importance of genetic diversity and introduction history for establishment ability.
Methods
Thirty-six tail tip biopsies, preserved in 95% ethanol, were taken from hand-caught and released lizards at the site of invasion, adjacent to the dock in the industrial port of Campden Park, on the south west coast of St Vincent, Lesser Antilles (Fig. 1a), following the conditions of permit and strictly adhering to the protocol of the Academic Ethics Committee (AEC). Locality positions were recorded using a handheld Garmin 60CS GPS. Samples were collected throughout the limited range of the introduced population (~400 m2). To locate the geographic origin of the introduced population, A. sagrei mtDNA sequences representing areas from across the native (74 populations) and introduced (29 populations) range were used for comparison with St Vincent samples. These mtDNA sequences were deposited in GenBank by Kolbe et al. (2004) accession numbers AY655172-484; DQ846752-771. Eight outgroup haplotypes were used comprising three congeneric Cuban natives; A. homolechis, A. bremeri and A. quadriocellifer (GenBank accession numbers AY655164-171).

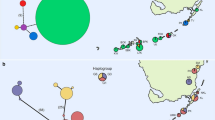
a Map showing geographic distribution between A. sagrei haplotypes within the SE Cuba, WC Florida, W Jamaica, St Vincent clade. Grey squares indicate haplotypes from Cuba, open squares indicate haplotypes from Florida/St Vincent, lined squares indicate haplotypes from Jamaica. 257 A. sagrei haplotypes from 74 native range populations and 77 haplotypes from 29 introduced populations from Kolbe et al. (2004) were included in this study (not marked on the map). b Network analysis showing genealogical relationship between A. sagrei haplotypes, shown in a, the number of nucleotide substitutions between haplotypes is shown by the number of dots connecting them in the network (Haplotype Identifiers: StV/TII = St Vincent/Tampa, Florida II; TI = Tampa, Florida I; StPI = St Petersburg, Florida I; StPII = St Petersburg, Florida II; JMI = Manchester, Jamaica I; JMII = Manchester, Jamaica II; JWI = Westmoreland, Jamaica I; JWII = Westmoreland, Jamaica II; PI = Portillo, Cuba I; P II = Portillo, Cuba II; NML I = N Media Luna, Cuba I; NML II = N Media Luna, Cuba II; NML III = N Media Luna, Cuba III)
Total genomic DNA was extracted using the DNeasy Tissue Kit (QIAGEN) following the manufacturer’s protocol for animal tissues. Approximately 1,200 bp of mtDNA, including NADH2 and two flanking tRNAs (tRNATrp tRNAAla) was amplified using the primers H5730 (Glor et al. 2004) and L4437b (Macey et al. 1997). The amplifications were performed in a total volume of 25 μl using 50 ng total genomic DNA, 20 μl (1.5 mM MgCl2) ReddyMix® PCR Master Mix (ABGene Surrey, UK) and 1 μM of each primer. PCR amplifications were performed using an MJ Research PTC-200 thermal cycler (Waltham, MA) using the following temperature cycling profile: a 15-min denaturation step at 95°C followed by 30 cycles of denaturation for 30 s at 94°C, annealing for 30 s at 50°C and extension for 1 min at 70°C. A final step at 72°C for 10 min followed the 30 cycles. Unincorportated deoxynucleotides and primers were removed using Promega 10× SAP and New England BioLabs Exonuclease I, following the manufacturer’s protocols. The forward and reverse primers were used for sequencing using dye-labelled terminators (ABI PRISM™ BigDye™ Terminator Cycle Sequencing Ready Reaction Kit) and subsequently run on an ABI Prism 377 DNA sequencer.
Sequences were aligned using CODONCODE ALIGNER 1.2 (CodonCode Co.) and replicated haplotypes were excluded from the analysis in order to reduce computational time. The substitutional model that best fit the data was assigned according to distance analyses performed using the log likelihood function of modeltest 3.0 (Posada and Crandall, 1998), and the chosen model was then used for subsequent distance-based analyses. McDonald and Kreitman’s (1991) test was used to evaluate the possibility of non-neutral evolution of the NADH2 gene (Ballard and Kreitman 1995). The genetic data analysis software packages PAUP × 4.0b10 (Swofford 1998) and MRBAYES version 3.1 (Huelsenbeck and Ronquist 2001) were used to reconstruct phylogenetic trees by maximum parsimony (MP) and Bayesian methods, respectively. Analyses included 334 unique haplotypes (excluding eight outgroups) from native and introduced populations across the native and invasive ranges, between one and ten unique haploypes per population. This strategy enabled all available information to contribute towards producing the most structurally informative phylogeny possible.
In all analyses, all sites were equally weighted. MP was performed using a random addition heuristic search with tree-bisection-reconnection with 1,000 random addition sequence replicates. Bootstrapping (1,000 replicates) was performed to obtain a relative measure of node support for the resulting MP tree (Felenstein 1985). The model of evolution used for Bayesian analysis was estimated by modeltest 3.0 and the parameter values were estimated by MRBAYES and fixed for the analysis. Four chains were run, with 2 million generations, sampling the chains every hundred generations. Time to stationarity was estimated by plotting tree log-likelihood score against generation number and determining the number of generations until the values reached an asymptote. The initial 25% of trees generated prior to stationarity were discarded as the “burn-in”. A majority rule consensus tree (‘Bayesian’ tree) was calculated from the posterior distribution of trees, and the posterior probabilities calculated as the percentage of samples recovering any particular clade (Huelsenbeck and Ronquist 2001). Three further independent Bayesian analyses were run to check for local optima.
A haplotype network was constructed using the network building software TCS (Clement et al. 2000) which uses a statistical parsimony procedure (Templeton et al. 1992). TCS produces a network linking different haplotypes only if they have a 95% probability of being justified by the parsimony criterion. Although network analysis is traditionally used to show genealogical relationships between haplotypes within populations (Templeton et al. 1992), it gives a useful visual indication of the number of steps (nucleotide differences) between haplotypes. Within-population nucleotide diversity (π) was calculated using MEGA3.1 (Kumar et al. 2004) for all populations sampled with more than one unique haplotype.
Results
A 1,194 bp fragment of mtDNA from a total of 342 sequences were used in the analysis. A single haplotype was identified from 36 sequenced St Vincent A. sagrei individuals. This haplotype was identical to a haplotype from Tampa, Florida, and the duplicate sequence was removed from further analyses. No insertions, deletions or stop codons were found in the data and the substitutional model of molecular evolution rejected the null hypothesis of neutral evolution for any comparison. Of the 1,194 sites, 666 were variable, 547 of which were parsimony-informative.
Bootstrapped MP and Bayesian analyses gave congruent tree topologies with similar, high bootstrap values, summarised in the Bayesian tree (Fig. 2). Both trees group St Vincent A. sagrei in a clade with 12 haplotypes from 6 localities across South East Cuba, West Central Florida and West Jamaica (shaded clade shown in Fig. 2; inset). Phylogenetic tree support was high for the clade (posterior probability 0.98, MP bootstrap support 81%, Fig. 2). Support for the 4 taxa clade grouping the St Vincent haplotype with Tampa and St Petersburg was high (posterior probability 1.00, MP bootstrap support 85%). The geographical distribution of A. sagrei haplotypes within the SE Cuba, WC Florida, W Jamaica clade is presented with geneological relationships among these haplotypes obtained from the network analysis (Fig. 1b). This clade was unconnected to other haplotypes because only connections with a probability higher than 95% are shown. Network analysis separates St Vincent/Tampa II from St Petersburg by 1 step, Tampa I by 4 steps, W Jamaica by 6 steps and from SE Cuba by 7 steps (Fig. 1b). Native populations have typically higher within-population nucleotide diversities, while introduced populations have typically lower diversities (Fig. 3). A notable exception is Florida, which contains among the oldest established of the introduced populations and has been colonised by multiple introductions from across the native range (Kolbe et al. 2004).

Bayesian phylogram of the NADH2 region sequence data, N = 342 haplotypes. Shaded clade is expanded in inset. Support for nodes are presented in the order Bayesian posterior probability value and maximum-parsimony bootstrap percentage. MtDNA haplotype data from Kolbe et al. (2004). Italics indicate colonised regions outside the native range

Histogram of within-population nucleotide diversity (π) for 1,194 bp within the mtDNA NADH2 region for all native and invasive populations included in the study, mtDNA haplotype data from Kolbe et al. (2004). Populations are grouped by country/state. Populations with no bar contain a single haplotype (4 populations from Cuba; 2 from Grand Cayman; Belize; 11 from Florida; 1 from Texas; 2 from Jamaica and St Vincent)
Discussion
Identical mtDNA haplotypes from St Vincent and Tampa II, FL supports a Tampa origin for the St Vincent population. Additionally, both MP and Bayesian tree topologies converge and group St Vincent into a well-supported clade with three haplotypes from Tampa and St Petersburg, FL, localities within small geographic distances (20 km). Network analyses group St Vincent, St Petersburg and Tampa together, separated from the next closest Jamaican haplotype by five steps, further supporting Tampa, FL as the source area for the St Vincent population. Commercial container imports have been routinely received at the Campden Park dock, St Vincent from the Northern Florida area during the period when the introduction occurred (F. Providence, personnel communication) while we are not aware of imports from any other of the native or introduced A. sagrei ranges, providing additional evidence for a Florida origin for introduced St Vincent A. sagrei.
The absence of the St Vincent/Tampa II haplotype from the native range haplotype set despite the use of data from a comprehensive study of intensive sampling effort (257 haplotypes from 74 populations from Kolbe et al. 2004), highlights a recurring problem when attempting to elucidate the origin of a new population of a widespread species. It is improbable that the haplotype would have been lost in the native range though drift, or formed in the invasive range through mutation since introduction to Florida in the late 1800’s (Williams 1969). The most probable explanation for the situation is that the haplotype exists in the native range but is absent from the sample set. This absence may be explained by the high genetic diversities within native range populations (Fig. 3; Table 1). Even thorough sampling will detect only a subset of the most frequently occurring haplotypes in these populations, with those at lower frequencies being overlooked. Following this logic, it is plausible, though perhaps less probable that the St Vincent population may originate from a source population in the native range which contains the St Vincent/Tampa II haplotype at a low frequency.
A recent study has also recognised that native population genetic diversity and sampling efficiency can substantially affect the accuracy of putative source population identification (Muirhead et al. 2008). In a simulation using published data sets, both the degree of genetic differentiation between source populations, and the number of source populations included in the study were found to significantly impact the accuracy of source population assignment. Furthermore, small sample sizes affected population assignment accuracy most when introduced populations comprised of dominant or fixed haplotypes that were at low frequencies in source populations (Muirhead et al. 2008). For this study, the sample size in the introduced St Vincent range was high (36 individuals in a single locality), and a large number of potential source populations were sampled (74 native populations and 29 populations across the introduced range). However, in accordance with Muirhead et al. (2008), because the haplotype was fixed in the introduced range and genetic differentiation between potential source populations was high (Fig. 3; Table 1), the estimation of the putative source population should be treated with some prudence.
Assuming a Tampa origin for A. sagrei in St Vincent, this recent importation represents a secondary introduction event, i.e. originating from an introduced rather than a native population. Other recent secondary introductions originating from Florida A. sagrei have been shown to retain a proportion of the genetic variation found within Floridian populations (Kolbe et al. 2004). In contrast we demonstrate that despite high within-population variation in the source population, Tampa (7.1% within-population nucleotide diversity, Table 1), the introduced population of A. sagrei in St Vincent, which contains a single mtDNA haplotype has experienced the most severe genetic depauparation of any of the documented global A. sagrei introductions. Such a complete absence of genetic diversity has rarely been documented in other introduced populations (although see Puillandre et al. 2008), although variation in more sensitive population genetic markers may represent severe founder effects (e.g. microstatellites, Hawley et al. 2006). For A. sagrei in St Vincent, the implied genetic founder effect is likely to be a result of colonisation history processes, directly via a demographic bottleneck, and augmented by the early stage of the St Vincent introduction, which was sampled before any subsequent, additional introductions may have had an opportunity to occur.
This study highlights the importance of determining source populations so that valid comparisons may be made with native populations. Such comparisons may be valuable in elucidating adaptive and plastic responses to environmental pressures, the influence that habitat preferences may have for population dynamics, potential impacts on native species and importantly, possible short- and long-term management strategies. For example, recent investigations have demonstrated that newly founded populations containing high levels of genetic diversity have shown rapid evolutionary responses to local conditions (Costa-Pierce 2003). In such cases, diversity often arises from the admixture of haplotypes originating from multiple introductions. Conversely, when a strong founder effect is combined with no admixture, the minimal amounts of available variance present are unlikely to enable adaptive genetic responses (Allendorf and Lundquist 2003), although some additive genetic variance may still be available to selection following demographic bottlenecks (e.g. via epistasis, Carson 1990).
The data presented here demonstrate, in contrast to previous genetic characterisation of introduced A. sagrei populations (Kolbe et al. 2004), that founder populations can exhibit extremely low haplotype diversity despite high source population genetic diversity, probably as a result of colonisation history. This study identifies a single mtDNA haplotype in a recent anthropogenically imported population of A. sagrei on St Vincent, identical to a haplotype from Tampa, FL, suggesting that Tampa is the geographical source for the introduction. However, it is also shown that multiple haplotypes with high nucleotide diversities within native range populations, combined with low frequencies (rareness) of introduced haplotypes in native ranges can impair attempts to identify source populations, even despite intensive sampling effort. Thus, both phylogenetic structure and within-population genetic diversities must be appreciated to identify the likely geographical source for introduced populations. Together with Muirhead et al. (2008) we recommend careful consideration of sampling design to avoid inaccurate assignment to putative source populations due to sampling errors.
References
Allendorf FW, Lundquist LL (2003) Population biology evolution and control of invasive species. Conserv Biol 17:24–30. doi:10.1046/j.1523-1739.2003.02365.x
Avise JC (1994) Molecular markers, natural history and evolution. Chapman and Hall, New York
Ballard JWO, Kreitman M (1995) Is mitochondrial DNA a strictly neutral marker? Trends Ecol Evol 10:485–488. doi:10.1016/S0169-5347(00)89195-8
Calsbeek R, Smith TB (2003) Ocean currents mediate evolution in island lizards. Nature 426:552–555. doi:10.1038/nature02143
Campbell TS (1996) Northern range expansion of the brown anole (Anolis sagrei) in Florida and Georgia. Herpetol Rev 27:155–157
Campbell TS (2003) The introduced brown anole (Anolis sagrei) occurs in every county in peninsular Florida. Herpetol Rev 34:173–174
Campbell TS, Echternacht AC (2003) Introduced species as moving targets: changes in body sizes of introduced lizards following experimental introductions and historical invasions. Biol Invasions 5:193–212. doi:10.1023/A:1026172314139
Carson HL (1990) Increased genetic variance after a population bottleneck. Trends Ecol Evol 5:228–230. doi:10.1016/0169-5347(90)90137-3
Clement M, Posada DA, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659. doi:10.1046/j.1365-294x.2000.01020.x
Costa-Pierce BA (2003) Rapid evolution of an established feral tilapia (Oreochromis spp): the need to incorporate invasion science into regulatory structures. Biol Invasions 5:71–84. doi:10.1023/A:1024094606326
Durka W, Bossdorf O, Prati D et al (2005) Molecular evidence for multiple introductions of Garlic mustard (Alliaria petiolata, Brassicaceae) to North America. Mol Ecol 14:1697–1706. doi:10.1111/j.1365-294X.2005.02521.x
Felenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evol Int J Org Evol 39:783–791. doi:10.2307/2408678
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to Conservation Genetics. Cambridge University Press, Cambridge
Glor RE, Gifford ME, Larson A et al (2004) Partial island submergence and speciation in an adaptive radiation: a multilocus analysis of the Cuban green anoles. Proc R Soc Lond B Biol Sci 271:2257–2265. doi:10.1098/rspb.2004.2819
Greene BT, Yorks DT, Parmerlee JS et al (2002) Discovery of Anolis sagrei in Grenada with comments on its potential impact on native anoles. Caribb J Sci 38:270–272
Hawley DM, Hanley D, Dhondt AA et al (2006) Molecular evidence for a founder effect in invasive house finch (Carpodacus mexicanus) populations experiencing an emergent disease epidemic. Mol Ecol 15:263–275. doi:10.1111/j.1365-294X.2005.02767.x
Huelsenbeck JP, Ronquist FR (2001) mrbayes: Bayesian inference in phylogeny. Bioinformatics 17:754–755. doi:10.1093/bioinformatics/17.8.754
Kolbe JJ, Glor RE, Rodrıguez Schettino L et al (2004) Genetic variation increases during biological invasion by a Cuban lizard. Nature 431:177–181. doi:10.1038/nature02807
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5:150–163. doi:10.1093/bib/5.2.150
Lee JC (1992) Anolis sagrei in Florida: phenetics of a colonising species I. meristic characters. Copeia 1985:182–194. doi:10.2307/1444808
Lee CE (2002) Evolutionary genetics of invasive species. Trends Ecol Evol 17:386–391. doi:10.1016/S0169-5347(02)02554-5
Macey JR, Larson A, Ananjeva NB et al (1997) Evolutionary shifts in three major structural features of the mitochondrial genome among Iguanian lizards. J Mol Evol 44:660–674. doi:10.1007/PL00006190
McDonald JH, Kreitman M (1991) Adaptive protein evolution at the adh locus in Drosophila. Nature 351:652–654. doi:10.1038/351652a0
McKeown S (1996) A field guide to reptiles and amphibians in the Hawaiian islands. Diamond Head Publishing Inc, Los Osos
Muirhead JR, Gray DK, Kelly D et al (2008) Identifying the source of species invasions: sampling intensity vs. genetic diversity. Mol Ecol 17:1020–1035. doi:10.1111/j.1365-294X.2008.03669.x
Norval G, Mao JJ, Chu HP et al (2002) A new record of an introduced species, the brown anole (Anolis sagrei) (Duméril & Bibron, 1837), in Taiwan. Zool Stud 41:332–336
Posada D, Crandall KA (1998) modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818. doi:10.1093/bioinformatics/14.9.817
Puillandre N, Dupas S, Dangles O et al (2008) Genetic bottleneck in invasive species: the potato tuber moth adds to the list. Biol Invasions 10:319–333. doi:10.1007/s10530-007-9132-y
Sakai AK, Allendorf FW, Holt JS et al (2001) The population biology of invasive species. Annu Rev Ecol Syst 32:305–332. doi:10.1146/annurev.ecolsys.32.081501.114037
Schloesser DW, Nakepa TF, Mackie GL (1996) Zebra Mussel infestation of unionid bivalves (Unionidae) in North America. Am Zool 36:300–310
Schoener TW, Gorman GC (1968) Some niche differences among three species of lesser Antillean anoles. Ecology 49:819–830. doi:10.2307/1936533
Shigesada N, Kawasaki K (1997) Biological Invasions: theory and practice. Oxford University Press, Oxford
Stenson AG, Thorpe RS, Malhotra A (2004) Evolutionary differentiation of bimaculatus group anoles based on analyses of mtDNA and microsatellite data. Mol Phylogenet Evol 32:1–10. doi:10.1016/j.ympev.2003.12.008
Suarez AV, Tsutsui ND (2008) The evolutionary consequences of biological invasions. Mol Ecol 17:351–360. doi:101111/j1365-294X200703456x
Swofford DL (1998) Phylogenetic analysis using parsimony (* and other Methods). Sinauer Associates, Sunderland
Templeton AR, Crandall KA, Sing CF (1992) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data III Cladogram estimation. Genetics 132:619–633
Williams EE (1969) The ecology of colonisation as seen in the zoogeography of anoline lizards on small islands. Q Rev Biol 44:345–389. doi:10.1086/406245
Acknowledgments
We would like to thank the St Vincent Forestry Department, in particular Fitzgerald Providence, for information and granting of permits necessary for field collection of material. Further thanks to Cathy Pook and Yann Surget-Groba for laboratory assistance, and Rob Ogden and an anonymous referee for valuable comments on earlier drafts of the manuscript. This work was funded by a NERC PhD studentship to JE, with CASE partner Wildlife DNA Services.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Eales, J., Thorpe, R.S. Revealing the geographic origin of an invasive lizard: the problem of native population genetic diversity. Biol Invasions 12, 77–86 (2010). https://doi.org/10.1007/s10530-009-9431-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-009-9431-6