
Abstract
Objectives
To develop a practical method to prepare tilianin by highly selective and efficient hydrolysis of the C-7 rhamnosyl group from linarin.
Results
Naringinase was utilized to selectively catalyze the formation of tilianin using linarin as the starting material. The reaction conditions, including temperature, pH, metal ions, substrate concentration and enzyme concentration, were optimized. At 60 °C, naringinase showed enhanced α-l-rhamnosidase activity while the β-d-glucosidase activity was abrogated. The addition of Mg2+, Fe2+ and Co2+ was also beneficial for selective biotransformation of linarin to tilianin. Under the optimized conditions (pH 7.0 at 60 °C), linarin could be nearly completely transformed to tilianin with excellent selectivity (>98.9 %), while that of the by-product acacetin was less than 1.1 %. In addition, the structure of target product tilianin was fully characterized by HR-MS and 1H-NMR.
Conclusion
A highly selective and efficient biotransformation of linarin to tilianin was developed by the proper control of incubation temperature, which enhanced the α-l-rhamnosidase activity of naringinase and blocked its β-d-glucosidase activity.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Tilianin (acacetin-7-o-β-d-glucoside), a rare flavonol glycoside, has received increasing attention for its various bioactivities including sedative, anti-hypertensive and anti-seizure effects, etc. (Zielinska and Matkowski 2014; Galvez et al. 2015). However, it is relatively expensive and hard to obtain, due to its low contents in plants (Zielinska and Matkowski 2014) and the difficulty in chemical synthesis (Liu et al. 2012). Tilianin is a hydrolytic product of linarin (acacetin-7-o-β-d-rutinoside), an abundant compound in numerous medical plants, such as Flos chrysanthemi indici, Buddleja officinalis, Cirsium setosum, Mentha arvensis, Buddleja davidii and Chrysanthemum boreale (Feng and Wang 2015; Nugroho et al. 2013). Compared with linarin, tilianin possesses a wide range of pharmacological activities including vasorelaxant, anti-atherogenic, anti-seizure effects, as well as potent therapeutic effects against angina pectoris and ischemia myocardial (Hernández-Abreu et al. 2013; Galvez et al. 2015; Zeng et al. 2016). Thus, it is necessary to develop an efficient and practical scheme to prepare tilianin using linarin as the starting material.
Linarin contains a rutinoside attached on the C-7 site of acacetin (see Fig. 2 below) and it could be transformed to tilianin (acacetin-7-o-β-d-glucoside) via selective hydrolysis of the rhamnosyl group. Although several enzymatic methods have been developed for selective biotransformation of flavonoid rutinoside to its glucoside (Wang et al. 2013), no practical method has been developed for highly efficient preparation of tilianin from linarin. There are two major challenges for highly efficient biotransformation of linarin to tilianin, one is its poor solubility (0.059 mg linarin/l water) of linarin in reaction system (Han et al. 2012), and another is the lack of enzyme(s) which can catalyze the biotransformation of linarin to tilianin with high selectivity.
Naringinase (EC 3.2.1.40), a glycosidase produced by several microorganisms (e.g., Penicillium sp.), displays both α-l-rhamnosidase and β-d-glucosidase activities (Puri 2012; Vila-Real et al. 2011). In the debittering process of citrus fruit, the α-l-rhamnosidase activity of naringinase could convert naringin to rhamnose and prunin, while prunin is subsequently hydrolyzed by the β-d-glucosidase subunit of naringinase to obtain the aglycone (Vila-Real et al. 2011). In the present study, naringinase was used for the formation of tilianin via selective hydrolysis of linarin for the first time. A series of factors influencing enzymatic hydrolysis of linarin, including reaction temperature, pH value, metal ions, substrate concentration and enzyme concentration, are investigated. Our results demonstrate that temperature can modulate the α-l-rhamnosidase and β-d-glucosidase activities of naringinase, and a highly selective and efficient biotransformation scheme was successfully developed to produce tilianin from linarin by naringinase via the proper control of incubation temperature. Under the optimized conditions, linarin could be nearly completely converted to tilianin with excellent selectivity of 98.9 %, which shows great potential for large-scale production.
Materials and methods
Biotransformation of linarin to tilianin catalyzed by naringinase
To remove the C-7 rhamnosyl group from linarin, the biotransformation was carried out in 50 mM citric acid/sodium citrate buffer, pH 7, (0.8 ml) by the commercial naringinase (0.1 mg naringinase/ml) from Penicillium decumbens. The stock solution of linarin was prepared in DMSO, and the final concentration of DMSO in the incubation system was 5 %. The reaction was incubated at various pH values, temperature, metal ions and chelator EDTA, linarin concentration, and different enzyme concentration while fixing other conditions in shaking water-bath. After incubation, the reaction was terminated by the addition of 1.6 ml ice-cold methanol, followed by centrifugation at 20,000×g for 20 min to obtain the supernatant for HPLC analysis. Blank controls without enzyme or substrate were carried out to ensure that the product formation was enzyme dependent.
Analysis of linarin and its products by LC-UV
The analysis of linarin and its hydrolytic products was performed by using an LC system with an ODS C18 column (150 × 4.6 mm, 5 μm; Kromasil) and eluting with acetonitrile (A) and water (B) at 1 ml/min, with a gradient program: 0–2.5 min, 65 % B-58 % B; 2.5–3 min, 58 % B-30 % B; 3–5.3 min, 30 % B-10 % B; 5.3–9.5 min, 10 % B; 9.5–10 min, balance to 65 % B, while column was kept at 40 °C. Linarin and the products were quantified according to their corresponding standard curves at 331 nm. For products analysis, 10 μl of the sample was injected into LC-UV.
Structure characterization of tilianin by high resolution mass spectrometry (HR-MS) and 1H-NMR
An Agilent 6540 UHD Accurate-Mass Q-TOF mass spectrometer (Agilent Technologies) via ESI ion source with Jet-Stream technology was used to analyze linarin and its hydrolytic products, and the MS spectra were recorded in positive ion mode from m/z 100–1000. The capillary voltage was set at 3.5 kV for positive ion detection. The flow rate and temperature of both sheath gas and drying gas were 8 l/min and 300 °C, respectively. The pressure of nebulizer gas was 35 psi. The fragmentor voltage is 50 V, while the Q-TOF acquisition rates were 1 and 1 Hz for full scanning and product ion scanning, respectively. The energies for collision induced dissociation (CID) experiments were set at 30 eV.
Tilianin was characterized by 1H-NMR. The biosynthesis reaction was carried out by incubating linarin (300 μM) with naringinase (0.1 mg naringinase/ml) in 50 ml citric acid/sodium citrate buffer (50 mM, pH 7.0) for 6 h at 60 °C, which was then terminated by adding 50 ml methanol. After centrifugation at 20,000×g for 20 min at 4 °C, the supernatant was concentrated and loaded on a SPE cartridge (C18, 1000 mg, Agela Technologies, Radnor, PA), which was then eluted and pooled in methanol. After vacuum concentration, the product was purified by HPLC. The structure of product was characterized on a Varian INOVA-400 NMR spectrometer (Varian, Palo Alto, CA). The purified product was dissolved in DMSO-d 6 (Euriso-Top, Saint-Aubin, France) for 1H-NMR analysis. Chemical shifts were given on δ scale and referenced to tetramethylsilane (TMS) at 0 ppm for 1H-NMR (400 MHz).
Results and discussion
Structural characterization of hydrolysate from linarin
The transformation of linarin by naringinase was conducted under the optimized conditions, and only one product peak was eluted at 4.8 min (Fig. 1). The hydrolytic product was then identified by HRMS in the positive ion mode, which was more sensitive than the negative ion mode (Supplementary Fig. 1; Supplementary Fig. 2). The molecular ion peak of this hydrolytic product is located at m/z 447.1251 (reducing m/z 146.0579 compared with linarin), corresponding to [M+H]+ of tilianin (C22H22O10).

The selective biotransformation of linarin to tilianin under the optimized conditions. The reaction was carried out by incubating linarin (178 mg/l) with naringinase (0.2 mg naringinase/ml) in 50 mM citric acid/sodium citrate buffer (pH 7.0) at 60 °C for 6 h
The hydrolytic product was further purified by LC, and then well characterized by 1H-NMR. The 1H-NMR spectrum data for linarin and tilianin were as follows:
Linarin, 1H-NMR (400 MHz, DMSO-d 6): δ 6.95 (1H, s, H-3), 6.46 (1H, d, J = 1.9 Hz, H-6), 6.80 (1H, d, J = 0.32 Hz, H-8), 8.07 (2H, d, J = 8.8 Hz, H-2′, 6′), 7.16 (2H, d, J = 8.8 Hz, H-3′, 5′), 3.87 (3H, s, 4′-OCH3), 5.08 (1H, d, J = 7.3 Hz, H-1″), 4.56 (1H, s, H-1‴), 3.18-5.46 (10H, m, sugar protons), 1.08 (3H, d, J = 6.1 Hz, H-6‴);
Hydrolytic product, 1H-NMR (400 MHz, DMSO-d 6): δ 6.96 (1H, s, H-3), 6.45 (1H, d, J = 0.48 Hz, H-6), 6.85 (1H, d, J = 0.44 Hz, H-8), 8.08 (2H, d, J = 8.9 Hz, H-2′, 6′), 7.14 (2H, d, J = 9.0 Hz, H-3′, 5′), 3.87 (3H, s, 4′-OCH3), 5.08 (1H, d, J = 7.3 Hz, H-1″), 3.20-5.42 (6H, m, H-2″-6″). These values were highly similar to the reported data (Zhang et al. 2012; Li et al. 2008).
On the basis of the above NMR spectral data, nearly all signals of the parent nucleus in both compounds appeared at the same position. Compared with linarin, the hydrolytic product lost the hydrogen signal at δ 4.56 and 1.08, but exhibited the same glucose signal at δ 5.08, which was considered as the deprivation of terminal rhamnose in 6″-substituted β-d-glucopyranoside. These results suggest that tilianin is effectively produced by the selective hydrolysis of linarin (Fig. 2).

Structures of linarin and tilianin
Optimization of the selectivity for the formation of tilianin
The effects of pH values on rhamnosyl group selectivity of naringinase are shown in Fig. 3a. There were large differences in the formation of tilianin and acacetin from linarin under various pH conditions. Linarin could be rapidly converted to acacetin from pH 3–5, with a high conversion rate (>99.5 %). However, two products including tilianin and acacetin could be detected from pH 5–8, while the formation of acacetin from linarin was significantly decreased from 99.5 to 6.3 % with higher pH values. The optimal pH for the formation of tilianin was 7 (Supplementary Fig. 3), with the highest conversion rate of 26 %.

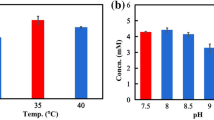
The effects of pH values and temperature on the formation of tilianin and acacetin from linarin by naringinase. The reactions were incubated in 50 mM citric acid/sodium citrate buffer for 6 h, with naringinase and linarin at 0.1 mg/ml and 30 mg/l, respectively. a The effects of pH values on the formation of tilianin (filled square) and acacetin (filled circle) at 37 °C; b The effects of temperature on the formation of tilianin (filled square) and acacetin (filled circle)
These results agree well with previous reports in which naringinase displayed both α-l-rhamnosidase and β-d-glucosidase activities, but the optimal pH for these two hydrolytic activities were distinct (Puri and Banerjee 2000; Ko et al. 2013). Thus, it was necessary to block the β-d-glucosidase activity of naringinase and enhance the conversion efficacy of tilianin from linarin via a more effective and economical way.
As shown in Fig. 3b, the effects of temperature on the rhamnosyl group selectivity of naringinase for the formation of tilianin were evaluated at pH 7.0. The different maximum conversions of linarin to tilianin and acacetin were observed at different temperatures. From 30 to 50 °C, there was significant enhancement in the formation of acacetin from 32 to 63 %, while tilianin was decreased to the lowest (<16 %). However, as the temperature increased, the formation of acacetin quickly declined to the trace amount (<0.1 %) above 50 °C, and linarin could be selectively converted to tilianin. The optimal temperature for the selective transformation of linarin to tilianin was 60 °C, where the formation of tilianin was up to 83 %.
These results indicated that α-l-rhamnosidase and β-d-glucosidase activities of naringinase had distinct thermal tolerance during the hydrolysis of linarin, and the β-d-glucosidase activity could be nearly abrogated over 60 °C. This was in good agreement with previous studies that α-l-rhamnosidase from Penicillium decumbens was optimal between 55 and 65 °C (Puri and Banerjee 2000; Magario et al. 2009). Similarly, α-l-rhamnosidase, from various species of Aspergillus, Rhizopus nigricans and Coniothrium stercorarium, was optimal above 60 °C (Yadav et al. 2000). However, compared with these purified enzymes, the easily available naringinase was more economical and practical for the selective formation of tilianin by the simple temperature control. Furthermore, this manufacturing technology is more technically feasible and easily achievable for scale-up processes. The high temperature increased solubility of natural flavonoids (Weignerova et al. 2012), especially linarin, (Han et al. 2012). Thus, the enzymatic reaction in the following experiments was carried out at 60 °C.
The effects of metal ions at 5 mM and EDTA on the selective formation of tilianin by naringinase are shown in Fig. 4. Mg2+, Fe2+, and Co2+ slightly enhanced the biotransformation of linarin to tilianin. However, Ca2+, Al3+, Fe3+, and Cu2+ increased the formation of acacetin indicating their unsuitability for the selective biotransformation of linarin to tilianin.

The effects of metal ions (5 mm) and EDTA on the formation of tilianin (filled column) and acacetin (blank column) from linarin by naringinase. The reactions were performed by incubating linarin (30 mg linarin/l) with naringinase (0.1 mg naringinase/ml) in 50 mM citric acid/sodium citrate buffer (pH 7.0) at 60 °C for 6 h, while the final concentration of metal ions was 5 mM. The control without metal ions or EDTA was carried out under the same reaction conditions
Optimization of the catalytic efficiency for the tilianin formation
The effect of substrate concentration on the formation of tilianin was evaluated from 12 to 178 mg linarin/l (Fig. 5). Linarin was selectively converted to tilianin, and the by-product, acacetin, was kept at a minimum conversion level lower than 0.15 %. Furthermore, the formation of tilianin from linarin rose from 76 to 89 % within the range of 12–148 mg linarin/l. Probably α-l-rhamnosidase activity of naringinase is not saturated at a low linarin concentration, and the reaction rate rose therefore rapidly as the substrate concentration increased. However, the upper limit of linarin solubility was 178 mg linarin/l and additional linarin caused precipitations. As a result, the optimal linarin concentration for the formation of tilianin is 178 mg linarin/l.

The effects of linarin concentration on the formation of tilianin (filled square) and acacetin (filled circle). The reactions were performed in 50 mM citric acid/sodium citrate buffer (pH 7.0) at 60 °C. Naringinase was at 0.1 mg/ml, while the substrate was from 12 to 178 mg/l
Under the above optimized reaction conditions (pH 7 at 60 °C), the formation of tilianin from linarin was investigated using 0.1–0.4 mg naringinase/ml (see Fig. 6). Following co-incubation with 0.3 or 0.4 mg naringinase/ml, the substrate (178 mg linarin/l) was nearly completely converted to tilianin within 4 h, with a very high conversion (>97.9 %). Using 0.2 mg naringinase/ml, the conversion rate of linarin gradually increased and finally reached 98.2 % following 8 h incubation; but at the lowest enzyme concentration (0.1 mg naringinase/ml), the final conversion rate of linarin was only 92.2 % following 12 h incubation. Overall, in the incubations above, the selectivity of the desired product (tilianin) was up to 98.9–99.8 %, while that of by-product (acacetin) was less than 1.1 %. In addition, the formation of tilianin by naringinase under the optimized conditions (pH 7 at 60 °C) was very fast and was not inhibited at high substrate concentration (Supplementary Fig. 4), indicating that α-l-rhamnosidase of naringinase exhibited a high reactivity towards linarin and thus it had great potential in large scale production of tilianin.

The effects of enzyme concentration on the formation of tilianin with 0.1–0.4 mg naringinase/ml. The enzymatic reactions were performed in the 50 mM citric acid/sodium citrate buffer (pH 7.0) at 60 °C, and with 178 mg linarin/l
Conclusion
Naringinase was used for the selective removal of the C-7 rhamnosyl group from linarin to produce tilianin at 60 °C. Under the optimized conditions, linarin could be nearly completely converted to tilianin with excellent selectivity of 98.9 % after incubation with naringinase, while that of the by-product acacetin was lower than 1.1 %. To the best of our knowledge, it is the first time to report a practical scheme for the highly selective and efficient hydrolysis of linarin to produce tilianin by using an easily available glycosidase.
References
Feng X, Wang X (2015) Linarin inhibits the acetylcholinesterase activity in vitro and ex-vivo. Iran J Pharm Res 14:949–954
Galvez J, Estrada-Reyes R, Benitez-King G et al (2015) Involvement of the GABAergic system in the neuroprotective and sedative effects of acacetin 7-O-glucoside in rodents. Restor Neurol Neurosci 33:683–700
Han Y, Xu J, Zhang TJ et al (2012) Determination of solubility and oil-water partition coefficient of linarin. Drug Eval Res 35:120–123
Hernández-Abreu O, Torres-Piedra M, García-Jiménez S et al (2013) Dose-dependent antihypertensive determination and toxicological studies of tilianin isolated from Agastache mexicana. J Ethnopharmacol 146:187–191
Ko JA, Ryu YB, Kwon HJ et al (2013) Characterization of a novel steviol-producing beta-glucosidase from Penicillium decumbens and optimal production of the steviol. Appl Microbiol Biotechnol 97:8151–8161
Li Y, Li J, Wang N et al (2008) Flavonoids and a new polyacetylene from Bidens parviflora Willd. Molecules 13:1931–1941
Liu JD, Chen L, Cai SL et al (2012) Semisynthesis of apigenin and acacetin-7-o-β-d-glycosides from naringin and their cytotoxic activities. Carbohydr Res 357:41–46
Magario I, Neumann A, Oliveros E et al (2009) Deactivation kinetics and response surface analysis of the stability of alpha-l-rhamnosidase from Penicillium decumbens. Appl Biochem Biotechnol 152:29–41
Nugroho A, Lim SC, Choi J et al (2013) Identification and quantification of the sedative and anticonvulsant flavone glycoside from Chrysanthemum boreale. Arch Pharm Res 36:51–60
Puri M (2012) Updates on naringinase: structural and biotechnological aspects. Appl Microb Biotechnol 93:49–60
Puri M, Banerjee UC (2000) Production, purification, and characterization of the debittering enzyme naringinase. Biotechnol Adv 18:207–217
Vila-Real H, Alfaia AJ, Rosa JN et al (2011) α-Rhamnosidase and β-glucosidase expressed by naringinase immobilized on new ionic liquid sol-gel matrices: activity and stability studies. J Biotechnol 152:147–158
Wang J, Sun GX, Yu L et al (2013) Enhancement of the selective enzymatic biotransformation of rutin to isoquercitrin using an ionic liquid as a co-solvent. Bioresour Technol 128:156–163
Weignerova L, Marhol P, Gerstorferova D et al (2012) Preparatory production of quercetin-3-beta-d-glucopyranoside using alkali-tolerant thermostable alpha-l-rhamnosidase from Aspergillus terreus. Bioresour Technol 115:222–227
Yadav V, Yadav PK, Yadav S et al (2000) α-l-Rhamnosidase: a review. Process Biochem 45:1226–1235
Zeng C, Jiang W, Tan M et al (2016) Optimization of the process variables of tilianin-loaded composite phospholipid liposomes based on response surface-central composite design and pharmacokinetic study. Eur J Pharm Sci 85:493–572
Zhang W, Xu J, Zhang L et al (2012) Flavonoids from the bran of Avena sativa. Chin J Nat Med 10:110–114
Zielinska S, Matkowski A (2014) Phytochemistry and bioactivity of aromatic and medicinal plants from the genus Agastache (Lamiaceae). Phytochem Rev 13:391–416
Acknowledgments
This work was supported by the National Basic Research Program of China (2013 CB531805), and National Natural Science Foundation of China (81573501, 81473181, 81302793, 81273590).
Supplementary information
Supplementary Figure 1— Representative HR-MS profile of linarin in citric acid/sodium citrate buffer (pH 7) Mass spectra of linarin (a) and and the fragmentation pathways of linarin (b) in positive ion mode .
Supplementary Figure 2—Representative HRMS profile of the hydrolytic product in citric acid/sodium citrate buffer (pH 7) by naringinase at 60 °C. Mass spectra of tilianin (a) and and the fragmentation pathways of tilianin (b) in positive ion mode.
Supplementary Figure 3—Representative HPLC-UV hydrolytic profile of linarin by naringinase in pH 4 and 7 citric acid/sodium citrate buffer. The reactions were incubated in 50mM citric acid/sodium citrate buffer for 6 h, while the final concentrations of naringinase and linarin were 0.1 mg/ml and 30 mg/l, respectively
Supplementary Figure 4—The enzyme kinetics analysis for the formation of tilianin from linarin by naringinase at the optimum conditions. To determine the kinetic behavior for the formation of tilianin, linarin (10–300 M) was incubated with naringinase (0.1 mg protein/ml) for 30 min in 50 mM citric acid/sodium citrate buffer (pH 7) at 60 °C.
Author information
Authors and Affiliations
Corresponding author
Additional information
Pan Cui and Tong-Yi Dou have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cui, P., Dou, TY., Li, SY. et al. Highly selective and efficient biotransformation of linarin to produce tilianin by naringinase. Biotechnol Lett 38, 1367–1373 (2016). https://doi.org/10.1007/s10529-016-2116-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-016-2116-1