
Abstract
The basic reactions of the clostridial 1-butanol biosynthesis pathway can be regarded to be the inverted reactions of the fatty acid β-oxidation pathway. A pathway for the biosynthesis of fuels and chemicals was recently engineered by combining enzymes from both aerobic and anaerobic fatty acid β-oxidation as well as enzymes from other metabolic pathways. In the current study, we demonstrate the inversion of the entire aerobic fatty acid β-oxidation cycle for 1-butanol biosynthesis. The constructed markerless and plasmidless Escherichia coli strain BOX-3 (MG1655 lacI Q attB-P trc-ideal-4-SDφ10-adhE(Glu568Lys) attB-P trc-ideal-4-SDφ10-atoB attB-P trc-ideal-4-SDφ10-fadB attB-P trc-ideal-4-SDφ10-fadE) synthesises 0.3–1 mg 1-butanol/l in the presence of the specific inducer. No 1-butanol production was detected in the absence of the inducer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
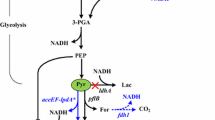
Many attempts have been made to construct non-native 1-butanol-producing microorganisms by cloning various heterologous genes in Escherichia coli (Atsumi et al. 2008a, 2008b; Inui et al. 2008; Nielsen et al. 2009), Bacillus subtilis (Nielsen et al. 2009), Pseudomonas putida (Nielsen et al. 2009), Saccharomyces cerevisiae (Steen et al. 2008), and Lactobacillus brevis (Berezina et al. 2010). While the construction of effective 1-butanol producers is still a challenge, the opportunity to create 1-butanol producers without using foreign genes is also of interest. The reactions of the fatty acid β-oxidation pathway, which is native in many organisms, and the main reactions of the clostridial 1-butanol biosynthesis pathway are the redox reaction sequences between metabolites with similar chemical structures (Fig. 1). We hypothesised that the reactions of the fatty acid β-oxidation pathway, like most redox reactions, could be reversed. The acyl-CoA derivatives, in particular butyryl-CoA, formed from acetyl-CoA could be reduced to their corresponding alcohols (in the simplest case, to 1-butanol) by endogenous CoA-dependent alcohol/aldehyde dehydrogenases.

Comparison of the widespread prokaryotic fatty acid β-oxidation pathway and 1-butanol biosynthesis pathway in Clostridium acetobutylicum. Enzymes of E. coli and C. acetobutylicum are indicated by their gene names: atoB acetyl-CoA C-acetyltransferase; fadA (fadI) acetyl-CoA C-acyltransferase; fadB (fadJ) 3-hydroxyacyl-CoA dehydrogenase/enoyl-CoA hydratase; fadE (ydiO) acyl-CoA dehydrogenase; thlA, thlB thiolase (acetyl-CoA acetyltransferase); hbd 3-hydroxybutyryl-CoA dehydrogenase; crt crotonase (3-hydroxybutyryl-CoA dehydratase); bcd butyryl-CoA dehydrogenase
Enzymes of the E. coli native fatty acid β-oxidation pathway can operate in a biosynthetic manner (Seregina et al. 2010). However, the corresponding strain TS325 was obtained by both classic genetic methods and mutagenesis stages that did not allow an estimation of the enzymes that formed the inverted β-oxidation pathway. In this study, we present evidence that 1-butanol can be synthesised in E. coli cells by reversed action of the aerobic fatty acid β-oxidation pathway enzymes.
Recently, the functional reversal of the fatty acid β-oxidation pathway for the synthesis of advanced chemicals (Dellomonaco et al. 2011) has been reported. However, the engineered pathway included not only separate enzymes of aerobic and anaerobic β-oxidation, but also enzymes of alternate metabolic pathways that have similar actions. Moreover, a significant production of target substances was achieved only by the inactivation of the main alcohol/aldehyde dehydrogenase AdhE and overexpression of key genes from plasmids.
In this study, the constructed markerless and plasmidless BOX-3 strain synthesises 1-butanol as a result of acetyl-CoA to butyryl-CoA conversion that is catalysed by the enzymes of the aerobic fatty acid β-oxidation pathway, which is followed by the reduction of butyryl-CoA to the corresponding alcohol by the main alcohol/aldehyde dehydrogenase AdhE. The BOX-3 strain contains only one chromosomal copy of each gene encoding the enzymes involved in the 1-butanol biosynthesis.
Materials and methods
Bacterial strains, plasmids and media
The bacterial strains and plasmids used in this study are listed in Supplementary Table 1. E. coli K-12 MG1655 (VKPM B-6195) was used as the parent strain for construction of all mutants described in this study.
All modifications in the E. coli MG1655 chromosome were obtained by the method of Datsenko and Wanner (2000). All of the primers that were used are listed in Supplementary Table 1. The modifications were obtained individually and then combined in the chromosome of the resulting strain BOX-3 (MG1655 lacI Q attB-P trc-ideal-4-SDφ10-adhE(Glu568Lys) attB-P trc-ideal-4-SDφ10-atoB attB-P trc-ideal-4-SDφ10-fadB attB-P trc-ideal-4-SDφ10-fadE) by sequential P1-mediated transductions. Detailed procedures for the strain construction are shown in the Supplementary Information.
Escherichia coli was grown in Luria–Bertani (LB), SOB, SOC and M9 media (Sambrook et al. 1989). Ampicillin (100 μg/ml), chloramphenicol (30 μg/ml) or kanamycin (50 μg/ml) was added when needed. IPTG, (1 mM) was used to induce the gene expression from the LacI-dependent promoter. Cells were grown aerobically on M9 plates (15 g agar/l) with ethanol (10 g/l) or sodium oleate (1 g/l) as sole carbon source. To improve sodium oleate solubility, Triton X-100 (4 g/l) was used.
Culture conditions
Semi-aerobic conditions
Cells of the BOX-3 strain and the control strain MG adhE568 were grown overnight in M9 medium containing 2 g glucose/l at 37°C. 1 ml of the overnight cultures was diluted 50 times with 49 ml M9 medium containing 10 g glucose/l. The cultures were grown in 750 ml flasks with ventilation plugs at 37°C on a shaker (250 rpm) for 6 h. Cell suspensions were centrifuged for 15 min at ~3,000×g and 4°C. The pellets were resuspended in 7 ml of M9 medium containing 20 g glucose/l and 10 g glycerol/l. The cell cultures were incubated for 24 h in 20 × 200 mm test tubes with ventilation plugs at 37°C on shaker (150 rpm) with or without the presence of 1 mM IPTG.
In addition, cells of the BOX-3 strain and the control strain MG adhE568 were grown overnight in LB medium at 37°C. 0.5 ml of the overnight cultures were diluted 100 times with the addition of 49.5 ml of LB medium. The cultures were grown in 750 ml flasks with ventilation plugs at 37°C on the shaker (250 rpm) for 3 h. The cell suspensions were centrifuged for 15 min at ~3,000×g and 4°C. The pellets were resuspended in 7 ml LB medium containing 20 g glucose/l. The cell cultures were incubated for 24 h in 20 × 200 mm test tubes with ventilation plugs at 37°C on shaker (150 rpm) in the presence of 1 mM IPTG.
Anaerobic conditions
The initial biomass accumulation in M9 and LB media was performed as described above. The cell pellets were resuspended in 15 ml of M9 or LB media containing 20 g glucose/l. The cell cultures were incubated for 24 h in 15 ml test tubes with non-ventilated plugs at 37°C on shaker (150 rpm) with or without 1 mM IPTG.
Analytical techniques
Fermentation samples were centrifuged for 10 min at 15,000×g. The supernatants were used for further analysis.
GC–MS was used to identify 1-butanol in culture media. The amounts of alcohols in culture media were determined by GC-FID. Concentrations of organic acids, glucose and glycerol in the culture media were measured by HPLC. Detailed procedures for metabolite detection and quantification are provided in the Supplementary information. Cell density was measured from the OD600 value; an OD600 of 1 = 400 mg dry wt per litre.
Results and discussion
Construction and characteristics of strains
The expression of genes encoding the enzymes of the fatty acid β-oxidation pathway in E. coli is repressed in the absence of fatty acids by the negative transcriptional regulator of fad regulon (the FadR protein) (Fujita et al. 2007). Moreover, the CoA-dependent alcohol/aldehyde dehydrogenase (AdhE), required for 1-butanol biosynthesis, is inactive in E. coli cells under aerobic conditions. Therefore, native regulatory regions of fadE (encoding acyl-CoA dehydrogenase), fadB (encoding 3-hydroxyacyl-CoA dehydrogenase end enoyl-CoA hydratase), atoB (encoding acetyl-CoA C-acetyltransferase), and adhE (encoding CoA-dependent alcohol/aldehyde dehydrogenase) genes were substituted by the artificial genetic element P trc-ideal-4-SDφ10, which contained a strong LacI-dependent promoter (P trc-ideal-4) and a Shine-Dalgarno sequence (SD sequence) of the φ10 gene from the phage T7. In the case of the adhE gene, a point mutation of G→A, leading to a Glu568Lys substitution in the protein product of the gene, was additionally introduced into position 1,702 of its coding region to obtain an aerotolerant mutant (Clark and Cronan 1980; Holland-Staley et al. 2000).
The BOX-3 strain was obtained by combining all modifications in the chromosome of the basic strain MG lacI Q. The engineered E. coli strain, MG lacI Q, was constructed by the introduction of a point mutation of CG→TA into the −35 region of P lacI promoter. This mutation is known to cause a lacI Q genotype that provides an effective repression of LacI-dependent promoters (Glascock and Weickert 1998).
The repression/induction efficiency of the expression of adhE, fadE, fadB and atoB genes, regulated by artificial genetic element P trc-ideal-4-SDφ10, and the functional activity of the corresponding proteins were verified by aerobically growing strains MG adhE568 and BOX-3 on M9 plates with ethanol or sodium oleate as a sole carbon source. While the strain MG adhE568 could aerobically grow on M9 plates that contained ethanol as a sole carbon source and IPTG as specific inducer, strain MG1655 did not grow either in the presence or absence of the inducer. Growth of the BOX-3 strain on M9 plates with sodium oleate as a sole carbon source was observed only in the presence of IPTG, while the growth of the strain MG1655 was independent of the presence of an inducer in the medium. Thus, the expression of the key genes of the fatty acid β-oxidation pathway in BOX-3 occurred only under induction; the corresponding enzymes were functionally active, thereby permitting the utilisation of fatty acids as the sole carbon source for cell growth.
1-Butanol biosynthesis by the BOX-3 strain under semi-aerobic conditions
Cultures of the BOX-3 strain and the control strain MG adhE568 were grown aerobically in M9 medium containing 10 g glucose/l as a sole carbon source. For 1-butanol fermentation, the biomass was cultivated semi-aerobically for 24 h in M9 medium containing 20 and 10 g glycerol/l, which was used as a source of additional NADH equivalents required for 1-butanol biosynthesis. In the absence of an inducer (IPTG), no 1-butanol was detected in the culture media of either the BOX-3 or MG adhE568 strain. In the presence of an inducer, 1-butanol was synthesised exclusively by the BOX-3 strain (Table 1A). The 1-butanol was identified in the culture media by GC–MS, and its amount was determined by gas chromatography using flame ionisation detector. As is shown in Table 1A, the amount of ethanol produced by the BOX-3 strain significantly exceeded the amount of 1-butanol. The low specificity of E. coli AdhE alcohol/aldehyde dehydrogenase to butyryl-CoA, as compared to acetyl-CoA, is responsible for the high ethanol/butanol ratio (Atsumi et al. 2008a). Moreover, only two NADHs are required to reduce acetyl-CoA to ethanol, while four NADHs are required to form 1-butanol from the same precursor. In this experiment (Table 1A), the fermentation products (such as lactate and succinate) were nearly absent in the culture medium, which indicates that the culture conditions of the strains were rather aerobic and that most of the NADH generated from glycolysis was reoxidized to NAD + through the respiratory selectron transport chain. The ethanol production by the BOX-3 and MG adhE568 strains in the absence of an inducer could be most likely due to the action of other enzymes that have similar activities, such as AdhP (EC 1.1.1.1), YqhD (EC 1.1.1.–), MhpF (EC 1.2.1.10) etc.
The ability of the BOX-3 strain to synthesise 1-butanol under semi-aerobic conditions was also shown when the strain was cultured in LB medium with 20 g glucose/l. As in the case of minimal medium, 1-butanol biosynthesis was observed only in the presence of an inducer (Table 1B). The control strain MG adhE568 was not able to produce 1-butanol either in the presence or the absence of an inducer. The amount of 1-butanol synthesised by the BOX-3 strain in the rich medium was higher the amounts seen in M9 medium, but the amount of ethanol was the same in both the rich and minimal media. Simultaneously, acetic acid accumulation in the rich medium was less. Thus, it is possible that the more effective redistribution of acetyl-CoA between competing reactions of ethanol biosynthesis and acetoacetyl-CoA formation led to a relatively higher 1-butanol biosynthesis by the BOX-3 strain.
1-Butanol biosynthesis by the BOX-3 strain under anaerobic conditions
The ability of BOX-3 to synthesise 1-butanol was also examined under anaerobic condition. The aerobically grown cells of strains BOX-3 and MG adhE568 were cultured in M9 or LB media supplemented with 20 g glucose/l for 24 h with or without 1 mM IPTG. As in semi-aerobic conditions, the 1-butanol synthesis was observed only in the BOX-3 strain upon induction of expression of fad regulon genes and alcohol/aldehyde dehydrogenase, irrespective of culture media (Table 3). Along with MG adhE568, the BOX-1 and BOX-2 strains, with altered regulation of an incomplete set of β-oxidation cycle genes, did not produce 1-butanol under any test condition (data not shown). The amounts of 1-butanol anaerobically produced by BOX-3 in rich medium were nearly identical to those synthesised under semi-aerobic conditions. On the other hand, the amount of 1-butanol produced by the BOX-3 strain in M9 media was higher under anaerobic conditions than in semi-aerobic conditions. Under anaerobic conditions, the main source of NADH in the minimal glucose media is via glycolysis, and the reducing equivalents are reoxidized in fermentative reactions. Thus, the redistribution of glycolytic NADH between fermentative pathways, instead of its oxidation through the respiratory electron transport chain, could increase the relative availability of reducing power for 1-butanol biosynthesis, thus causing the higher anaerobic production of 1-butanol by the BOX-3 strain in M9 media. Nevertheless, the amount of 1-butanol anaerobically synthesised by the BOX-3 strain remained low. The lack of NADH equivalents required for redox-balanced 1-butanol biosynthesis is responsible for this phenomenon. In the presence of O2, the NADH deficiency was caused by intensive oxidation; under anaerobic conditions, the NADH deficiency was caused by the insufficient generation of reducing equivalents. In the absence of O2, the formation of 2 mol acetyl-CoA is accompanied by the generation of 2 mol glycolytic NADH, instead of the 4 mol that are required for 1-butanol biosynthesis. Moreover, in the case of the BOX-3 strain, the NADH availability for anaerobic production of 1-butanol decreases, due to the activation of lactic acid biosynthesis. The lactate formation competes with 1-butanol biosynthesis for both NADH and its direct metabolic precursor, pyruvate. The pathways that compete with 1-butanol biosynthesis for both carbon and reducing power (acetate, lactate and succinate biosynthesis) are responsible for the utilisation of about 80% of the glucose consumed by the strain (Table 2A).
The results presented above have demonstrated that 1-butanol biosynthesis via the inverted aerobic fatty acid β-oxidation pathway is possible even at conditions that are far from optimal. We did not delete the host pathways that compete with 1-butanol biosynthesis for reducing equivalents and carbon sources. However, it has been shown that the inactivation of the competitive reactions in recombinant E. coli strains harbouring clostridial 1-butanol biosynthesis genes on multi-copy plasmids can increase the production of 1-butanol from 1 mg/l (13 μM) to 0.5 g/l (6.7 mM) (Donaldson et al. 2006; Atsumi et al. 2008a).
The driving force required for efficient 1-butanol production includes the balanced activities of 1-butanol synthesising enzymes and the redox status of the producer (Bond-Watts et al. 2011; Shen et al. 2011). This concept for the construction of heterological recombinant 1-butanol producers can be applied to the optimisation of the inverted fatty acid β-oxidation pathway for 1-butanol production. The basic strain RB01 (MG1655 fadR atoC(c) ∆arcA crp*), described by Dellomonaco et al. (2011), synthesised none of the products that can be derived from fatty acid β-oxidation intermediates; the 1-butanol production was achieved only after the inactivation of fermentative pathways by deleting adhE, pta and frdAB genes and overexpressing the yqhD gene, which codes for NADP-depended aldehyde dehydrogenase in the plasmid. In the present study, the inversion of fatty acid β-oxidation was achieved in the BOX-3 strain by induced expression of chromosomal copies of only aerobic fad regulon and adhE genes.
Conclusions
We have demonstrated the inversion of the entire aerobic fatty acid β-oxidation cycle in the biosynthetic direction. The constructed markerless and plasmidless strain BOX-3 synthesises 1-butanol as a result of acetyl-CoA to butyryl-CoA conversion via the inverted aerobic fatty acid β-oxidation pathway, followed by the reduction of butyryl-CoA to the corresponding alcohol by native alcohol/aldehyde dehydrogenase. This statement is based on the following results: (1) the only difference between the butanol-producing BOX-3 strain and the control strain MG adhE568 is that the native regulatory regions of three genes coding for fatty acid β-oxidation enzymes were substituted in the BOX-3 strain by the artificial regulatory elements; (2) the BOX-3 strain synthesises 1-butanol only upon the induction of the expression of these three genes, which are under the control of the artificial regulatory elements; and (3) the induction of only alcohol/aldehyde dehydrogenase expression is not sufficient for 1-butanol biosynthesis in the control strain MG adhE568.
References
Atsumi S, Cann AF, Connor MR, Shen CR, Smith KM, Brynildsen MP, Chou KJ, Hanai T, Liao JC (2008a) Metabolic engineering of Escherichia coli for 1-butanol production. Metab Eng 10:305–311
Atsumi S, Hanai T, Liao JC (2008b) Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature 451:86–99
Berezina OV, Zakharova NV, Brandt A, Yarotsky SV, Schwarz WH, Zverlov VV (2010) Reconstructing the clostridial n-butanol metabolic pathway in Lactobacillus brevis. Appl Microbiol Biotechnol 87:635–646
Bond-Watts BB, Bellerose RJ, Chang MC (2011) Enzyme mechanism as a kinetic control element for designing synthetic biofuel pathways. Nat Chem Biol 7:222–227
Clark DP, Cronan JE Jr (1980) Acetaldehyde-coenzyme A dehydrogenase of Escherichia coli. J Bacteriol 144:179–184
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645
Dellomonaco C, Clomburg JM, Miller EN, Gonzalez R (2011) Engineered reversal of the β-oxidation cycle for the synthesis of fuels and hemicals. Nature. doi:10.1038/nature10333
Donaldson GK, Huang LL, Maggio-Hall LA, Nagarajan V, Nakamura CE, Suh W (2006) Fermentative production of four carbon alcohols. International patent application PCT/US2006/038007, 28 Sept 2006
Fujita Y, Matsuoka H, Hirooka K (2007) Regulation of fatty acid metabolism in bacteria. Mol Microbiol 66:829–839
Glascock CB, Weickert MJ (1998) Using chromosomal lacI q1 to control expression of genes on high-copy-number plasamids in Escherichia coli. Gene 223:221–231
Gulevich AYu, Skorokhodova AYu, Ermishev VYu, Krylov AA, Minaeva NI, Polonskaya ZM, Zimenkov DV, Biryukova IV, Mashko SV (2009) A new method for the construction of translationally coupled operons in a bacterial chromosome. Mol Biol (Mosk) 43:505–514
Holland-Staley CA, Lee K, Clark DP, Cunningham PR (2000) Aerobic activity of Escherichia coli alcohol dehydrogenase is determined by a single amino acid. J Bacteriol 182:6049–6054
Inui M, Suda M, Kimura S, Yasuda K, Suzuki H, Toda H, Yamamoto S, Okino S, Suzuki N, Yukawa H (2008) Expression of Clostridium acetobutylicum butanol synthetic genes in Escherichia coli. Appl Microbiol Biotechnol 77:1305–1316
Katashkina JI, Skorokhodova AY, Zimenkov DV, Gulevich AY, Minaeva NI, Doroshenko VG, Biryukova IV, Mashko SV (2005) Tuning the expression level of a gene located on a bacterial chromosome. Mol Biol (Mosk) 39:719–726
Nielsen DR, Leonard E, Yoon SH, Tseng HC, Yuan C, Prather KL (2009) Engineering alternative butanol production platforms in heterologous bacteria. Metab Eng 11:262–273
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, second ed. Cold Spring Harbor Press, New York
Seregina TA, Shakulov RS, Debabov VG, Mironov AS (2010) Construction of a butyrate-producing E. coli strain without the use of heterologous genes. Appl Biochem Microbiol 46:745–754
Shen CR, Lan EI, Dekishima Y, Baez A, Cho KM, Liao JC (2011) Driving forces enable high-titer anaerobic 1-butanol synthesis in Escherichia coli. Appl Environ Microbiol 77:2905–2915
Skorokhodova AYu, Zimenkov DV, Gilevich AYu, Minaeva NI, Biriukova IV, Mashko SV (2006) Insertion of the symmetrical Olac-ideal between the “35” and “10” regions of the hybrid Ptrc/Olac promoter leads to a significant increase in the efficiency of the LacI-driven repression. Biotechnol Rus 3:1–14
Steen EJ, Chan R, Prasad N, Myers S, Petzold CJ, Redding A, Ouellet M, Keasling JD (2008) Metabolic engineering of Saccharomyces cerevisiae for the production of n-butanol. Microb Cell Fact 7:36
Acknowledgments
This work was supported by a grant from the Russian Foundation for Basic Research (10-04-00766-a). We thank Dr. Svetlana Antonova for carrying out the HPLC analysis. We are also grateful to Dr. Nikolay Ikonnikov for conducting the preliminary GC analysis.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Gulevich, A.Y., Skorokhodova, A.Y., Sukhozhenko, A.V. et al. Metabolic engineering of Escherichia coli for 1-butanol biosynthesis through the inverted aerobic fatty acid β-oxidation pathway. Biotechnol Lett 34, 463–469 (2012). https://doi.org/10.1007/s10529-011-0797-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-011-0797-z