
Abstract
A putative l-rhamnose isomerase (RhaA) from Thermotoga maritima was purified with a specific activity of 55 U/mg by His-Trap affinity chromatography. The native enzyme was estimated as a 46 kDa tetramer by gel filtration chromatography. The half-lives of the enzyme at 75, 80, 85, 90 and 95°C were 773, 347, 187, 118, and 65 h, respectively, indicating that it is the most thermostable of all RhaAs. Under the optimum conditions of pH 8.0, 85°C, and 1 mM Mn2+, RhaA with 100 U enzyme/ml converted 500 l-xylulose/l to 225 g/l l-lyxose after 3 h, and converted 500 l-fructose/l to 175 g/l l-mannose after 5 h.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
l-Rhamnose isomerase (RhaA, EC 5.3.1.14), reversibly catalyzes the isomerization of l-rhamnose to l-rhamnulose, participates in sugar metabolism such as mannose and fructose (Moralejo et al. 1993; Kanehisa and Goto 2000), and exists in various microorganisms (Domagk and Zech 1963; Izumori et al. 1976; Oudega et al. 1997). RhaAs from Escherichia coli, Pseudomonas stutzeri, and Bacillus pallidus have been characterized for conversion of monosaccharides (Badia et al. 1991; Leang et al. 2004b, c; Poonperm et al. 2007b). Thermostable enzymes have several process advantages for conversion of monosaccharides, such as high reaction velocities, resistance to chemical denaturation, reduced risk of contamination, and high substrate solubility (Bruce et al. 1991). However, the characterization of a thermostable RhaA has not yet been reported.
Rare l-form monosaccharides have recently attracted attention as potential starting materials for the synthesis of many pharmaceutical compounds (Doong et al. 1991). Their production has been studied using only a few enzymes and microorganisms (Itoh and Izumori 1996; Bhuiyan et al. 1999; Poonperm et al. 2007a; Rao et al. 2008) because most enzymes and microorganisms exhibit little activity for rare l-form monosaccharides. Therefore, the screening of enzymes that isomerize rare l-form monosaccharides is desired.
In the present study, to obtain an enzyme that produces rare l-form monosaccharides, the gene encoding a putative protein from the hyperthermophilic eubacterium Thermotoga maritima (Nelson et al. 1999; Conners et al. 2005) was cloned and expressed in E. coli. Evaluation of the substrate specificity of the expressed enzyme identified it as a RhaA. The enzyme exhibited high activity for l-form monosaccharides such as l-lyxose, l-xylulose, l-mannose, and l-fructose. This report describes the evaluation of this enzyme in terms of its potential for high-level production of l-lyxose and l-mannose.
Materials and methods
Bacterial strains, plasmid, and culture conditions
Genomic DNA from T. maritima ATCC 43589 D-5, E. coli ER2566, and pET-24a (+) were used as the source of RhaA gene, host cells, and expression vector, respectively. The recombinant E. coli for protein expression were cultivated with shaking at 200 rpm in a 2,000 ml flask containing 500 ml Luria–Bertani (LB) medium at 37°C with 20 μg kanamycin/ml until the OD600 reached 0.6. IPTG was added to 0.1 mM to induce enzyme expression and then the culture was grown at 16°C for 16 h.
Gene cloning
The gene encoding a putative protein was amplified by PCR using T. maritima genomic DNA as a template. The sequence of the oligonucleotide primers used for gene cloning was based on the DNA sequence of the protein from T. maritima MSB8 (GenBank accession number NP_228877). Forward (5′-AACATATGATAAACATGGAAAGGATT-3′) and reverse primers (5′-TTCTCGAGTCATCGTCTTCTTCTCCTTCT-3′) were designed to introduce the NdeI and XhoI restriction sites (underlined) and were synthesized by Bioneer (Daejon, Korea). The amplified DNA fragment obtained by PCR was purified and inserted into the pGEM-T easy vector (Promega, Madison, WI). E. coli Top10 strain was transformed with the ligation mixture and plated on LB agar containing 50 μg ampicillin/ml, 0.1 mM IPTG and 80 μg X-gal/ml. Ampicillin-resistant white colonies were selected, and plasmid DNA from these transformants was isolated using a plasmid purification kit (Solgent, Daejon, Korea). The NdeI-XhoI fragment from the T-vector containing the gene encoding RhaA was subcloned into the same sites of pET-24a (+) plasmid and the resulting plasmid was obtained. The plasmid was transformed into E. coli ER2566 strain and grown on LB medium containing 20 μg kanamycin/ml. The expression of the gene encoding RhaA was analyzed by both SDS-PAGE and assay of enzyme activity.
Enzyme purification
The cells were harvested from culture broth by centrifugation at 6,000×g for 20 min at 4°C, washed twice with 0.85% (w/v) NaCl, and resuspended in lysis buffer (pH 8.0) containing 50 mM NaH2PO4 and 300 mM NaCl with 1 mg lysozyme/ml. The resuspended cells were disrupted on ice using a sonicator. The supernatant of disrupted cell was applied onto a His-Trap HP chromatography column (Amersham Biosciences, Uppsala, Sweden) and eluted with a linear gradient from 10 to 250 mM imidazole at 1 ml min−1. The active fraction was collected and dialyzed against 50 mM N-(2-hydroxyethyl)piperazine-N′-(3-propanesulfonic acid) (EPPS) buffer (pH 8.0). After dialysis, the resulting solution was used as the purified enzyme.
Effects of metal ions, pH, and temperature
Unless otherwise stated, the reaction was performed in 50 mM EPPS buffer (pH 8.0) containing 10 mM l-rhamnose and 2.75 U enzyme/ml in the presence of 1 mM Mn2+ at 85°C for 10 min. To investigate the effect of metal ions on the activity of RhaA, the enzyme activity was measured after treatment with EDTA at 60°C for 1 h or after adding 1 mM of each metal ion, such as BaCl2, CaCl2, CoSO4, CuSO4, FeSO4, MgCl2, MnSO4, NiSO4, or ZnSO4 to EDTA-treated enzyme. To find the maximum activity of the enzyme, pH was varied from 6.5 to 8.5 using 50 mM PIPES buffer (pH 6.5–7.5) and 50 mM EPPS buffer (pH 7.5–8.5) at 85°C, and then at 75–95°C at pH 8.0. One unit of enzyme activity was defined as the amount of enzyme required to produce 1 μmol l-rhamnulose from l-rhamnose per min at 85°C and pH 8.0.
Determination of equilibrium ratio
The equilibrium ratio of l-lyxose (or l-mannose) to l-xylulose (or l-frucose) was determined as the average of the ratios obtained after incubation for 12 h at initial ratios of 0:100, 50:50, and 100:0. The reactions were performed at 85°C in 50 mM EPPS buffer (pH 8.0) containing 1 mM monosaccharides and 100 U enzyme/ml.
Analytical methods
The concentration of monosaccharides was determined using a Bio-LC system (Dionex ICS-3000, Sunnyvale, CA) with an electrochemical detector and a CarboPac PAI column. The column was eluted at 30°C with 200 mM NaOH at 1 ml/min. The concentrations of sugar phosphates were determined by the same system. The column was eluted at 30°C with a sodium-acetate gradient of 75 mM NaOH and 75 mM NaOH/500 mM sodium-acetate. The gradient was increased to 100 mM between 0 and 35 min, to 150 mM between 35 and 38 min, to 350 mM between 38 and 65 min and then to 500 mM for 75 min (Groussac et al. 2000). The flow rate was 1 ml/min.
Results and discussion
Gene cloning, purification, and molecular mass determination of the putative protein from T. maritima
The gene (1,152 bp) encoding the putative protein from T. maritima had the same sequence as a gene reported in GenBank (accession number NP_228877), was cloned and expressed in E. coli. The amino acid sequence of the protein from S. pneumoniae showed 18, 37, and 12% identity with those of RhaAs from E. coli, P. stutzeri, and B. pallidus, respectively. However, the active site residues of Trp 185, Glu226, Lys228, Asp258, His261, His286, Asp294, Asp296, and Asp326 in E. coli RhaA were absolutely conserved in those of the protein from T. maritima (Poonperm et al. 2007b). These results strongly suggest that the protein isolated from T. maritima was a RhaA.
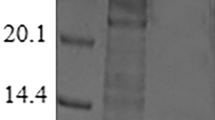
The enzyme was purified as a soluble protein from crude extract by His-Trap HP chromatography with a final purification of 23-fold, a yield of 42%, and a specific activity of 55 U/mg. The molecular mass of the purified enzyme from T. maritima, determined by SDS-PAGE, was approximately 46 kDa (Fig. 1), which is consistent with the calculated value of 45,527 Da with Compute pI/Mw software based on the 389 amino acid residues including 6 His residues. The native enzyme was estimated as a tetramer with a molecular mass of 184 kDa as determined by gel filtration chromatography using a Sephacryl S-300 HR 16/60 column. The column was calibrated with α-amylase from sweet potato (200 kDa), alcohol dehydrogenase from yeast (150 kDa), albumin (66 kDa), and carbonic anhydrase from bovine (29 kDa) as reference proteins.

SDS-PAGE analysis of RhaA from T. maritima at each purification step. Lane 1, prestained marker proteins (72, 55, 43, 34, and 26 kDa); lane 2, His-Trap HP column product (purified enzyme)
Substrate specificity of the putative protein from T. maritima for monosaccharides and aldose phosphates
The specific activity of the putative protein from T. maritima was investigated with the d-and l-forms of all pentoses and hexoses and with aldose phosphates, including d-ribose-5-phosphate, d-glucose-6-phosphate, and d-mannose-6-phosphate (Table 1). Among aldose substrates, the highest specific activity was observed with l-rhamnose, followed by l-lyxose, l-mannose, d-allose, d-gulose, and d-ribose. Among ketose substrates, the highest specific activity was observed with l-rhamnulose, followed by l-xylulose, d-ribulose, l-fructose, d-psicose, and d-sorbose. No activity was observed with aldose phosphates, including d-ribose 5-phosphate, d-glucose 6-phosphate and d-mannose 6-phosphate. The substrate specificities of RhaAs from E. coli, P. stutzeri and B. pallidus have been previously reported to follow the order l-rhamnose > l-lyxose > l-mannose among aldose substrates, the order l-rhamnose > l-lyxose > l-mannose > d-ribose > d-allose, and the order l-rhamnose > l-lyxose > d-ribose > l-mannose > d-allose, respectively (Poonperm et al. 2007b). RhaA from P. stutzeri converts one substrate to two different products by a two-step isomerization reaction. However, the enzyme has some disadvantages including by-product formation and low yields for the production of specific monosaccharides. In contrast, the putative protein from T. maritima isomerizes one product from one substrate. This putative protein is useful in producing specific monosaccharides and high yield is achieved without by-product formation.
The kinetic parameters of the enzyme for l-rhamnose, l-lyxose, l-mannose, l-xylulose, and l-fructose are shown in Table 2. The K m for l-rhamnose was 1.9-fold higher than for l-lyxose. The k cat for l-rhamnose was 3.2-fold higher than that for l-lyxose. As a result, the k cat/K m for l-rhamnose was 5.9-fold higher for l-lyxose. The specific activity and k cat/K m were highest for l-rhamnose among substrates, indicating that the sugar isomerase from T. maritima is a RhaA.
Effects of metal ions, pH, and temperature on the activity of RhaA from T. maritima
RhaAs from E. coli, P. stutzeri and B. pallidus have been reported as metal-dependent enzymes and their activities were the highest with Mn2+ (Badia et al. 1991; Leang et al. 2004c; Poonperm et al. 2007b). Of the metal ions tested, Mn2+ was the most effective for the isomerization of l-rhamnose by RhaA from T. maritima, resulting in 6.7-fold increase activity relative to no treatment. The optimal Mn2+ concentration was 1 mM (data not shown). Consequently, all subsequent experiments were performed in the presence of 1 mM Mn2+.
Maximum enzyme activity of RhaA from T. maritima was at pH 8.0 and 85°C (data not shown). The thermostability was examined by measuring the activity from 60 to 80°C. Thermal inactivation of the putative protein from T. maritima followed first-order kinetics and the half-life of the enzyme at 75, 80, 85, 90, and 95°C were 773, 347, 187, 118, and 65 h, respectively (Fig. 2).

Thermal inactivation of RhaA from T. maritima of 75 (filled circle), 80 (open square), 85 (filled square), 90 (open circle), and 95°C (filled triangle). For investigating thermostability, the enzymes were incubated at temperatures ranging from 75 to 95°C for varying periods of time. A sample was withdrawn at each time interval and was assayed for the remaining enzyme activity in 50 mM EPPS buffer (pH 8.0) containing 10 mM l-rhamnose and 1 mM Mn2+ at 85°C for 10 min. The experimental data for thermal deactivation of enzyme were fitted to a first order curve and the half-lives of the enzyme calculated using Sigma plot 9.0 software (Systat Software, San Jose, CA, USA). The relative activity of 100% was 2.75 U enzyme/ml. Data represent the means of three experiments and error bars represent standard deviation
The optimum temperatures and half-lives of RhaAs from the mesophiles E. coli, P. stutzeri, and B. pallidus are 60, 60, and 65°C, respectively, and as 10 min at 50°C, 10 min at 60°C, and 60 min at 60°C, respectively (Badia et al. 1991; Leang et al. 2004c; Poonperm et al. 2007b). However, RhaA from the hyperthermophile T. maritima examined in this study has an optimum of 85°C and a half-life of 773 h at 75°C, which is 773 times higher than that from B. pallidus RhaA at 60°C. In terms of the optimum temperature and thermal inactivation, RhaA of T. maritima is the most thermostable of the RhaAs reported to date.
Production of l-lyxose and l-mannose by RhaA from T. maritima
The equilibrium ratio between l-lyxose and l-xylulose was 45:55, while that between l-mannose and l-fructose was 35:65. The enzyme concentrations for the production of l-lyxose from 500 g l-xylulose/l and l-mannose from 500 g l-fructose/l were varied from 20 to 120 U enzyme/ml. The production (or conversion yield) of l-lyxose and l-mannose increased with increasing the concentrations of enzyme until reaching a plateau at 100 U enzyme/ml. The optimum concentration of the enzyme for effective production of l-lyxose and l-mannose was 100 U enzyme/ml (Fig. 3).

Effect of enzyme activity on the production of l-lyxose and l-mannose by RhaA from T. maritima. Conversion of l-lyxose (filled circle) from l-xylulose and Conversion of l-mannose (filled square) from l-fructose. The enzyme activity was varied from 20 to 120 U enzyme/ml for the production of l-lyxose and l-mannose. The reactions were performed in 50 mM EPPS buffer (pH 8.0) containing 500 g l-xylulose/l or 500 g l-fructose/l and 1 mM Mn2+ at 85°C for 5 h. Data represent the means of three experiments and error bars represent standard deviation
The conversion yields of l-lyxose and l-mannose were assessed by varying l-xylulose and l-fructose concentrations, respectively, from 50 to 500 g/l after 5 h. Increases in substrate concentration led to proportional increases in the production of l-lyxose and l-mannose while the conversion yield of l-lyxose from l-xylulose and that of l-mannose from l-fructose were almost constant as 45 and 35%, respectively, regardless of substrate concentration.
The production of l-lyxose and l-mannose by RhaA from T. maritima was performed in 500 g l-xylulose/l and 500 g l-fructose/l, respectively, with 100 U enzyme/ml for 5 h (Fig. 4). The concentrations of l-lyxose and l-mannose reached maximum values of 225 g l-lyxose/l at 3 h and 175 g l-mannose/l after 4 h, respectively. The conversion yields of l-lyxose from l-xylulose and productivity of l-lyxose were 45% and 75 g l−1 h−1, respectively, while the conversion yield of l-mannose from l-fructose and productivity of l-mannose were 35% and 35 g l−1 h−1, respectively. P. stutzeri RhaA produced 4.1 g l-lyxose/l from 19.2 g l-xylulose/l for 47 h with a conversion yield of 21% and a productivity of 0.09 g l−1 h−1 (Granstrom et al. 2005), and the enzyme produced 30 g l-mannose/l from 100 l-fructose/l for 6 h with a conversion yield of 30% and a productivity of 5 g l−1 h−1 (Bhuiyan et al. 1997). Compared to the concentration and productivity observed with RhaA from P. stutzeri, those of l-lyxose observed with RhaA from T. maritima in the present study were 55- and 833-fold higher and those of l-mannose were 5.8- and 7.0-fold higher. As a result, the highest ever reported concentrations and productivities of l-lyxose and l-mannose were obtained.

Production of l-lyxose and l-mannose by RhaA from T. maritima. a Production of l-lyxose (filled circle) from l-xylulose (open circle). b Production of l-mannose (filled square) from l-fructose (open square). The reactions were performed in 50 mM EPPS buffer (pH 8.0) containing 500 g l-xylulose/l or 500 g l-talose/l, 100 U ml−1 enzyme, and 1 mM Mn2+ at 85°C for 5 h. Data represent the means of three experiments and error bars represent standard deviation
For the separation of the product monosaccharide from the reaction mixture, the sample was treated with activated charcoal and filtrated to remove the charcoal. The filtrate was deionized by passing ion exchange resins such as Diaion SK1B (H+ form) and Amberlite IRA-411 (CO3 2− form). The deionized sample is applied to Dowex 50 W-X2 (Ca2+ form) or Amberlite CR-1220 (Ca2+ form) resin and the bound monosaccharide is eluted. The fractions containing the product were pooled and evaporated. The concentrated sample solidified into the pure monosaccharide with addition of a small amount of the product monosaccharide (Itoh et al. 1995; Leang et al. 2004a). Using these methods, l-lyxose or l-mannose produced in the present study can be purified as the pure monosaccharide from the reaction mixture.
l-Lyxose is a useful l-form monosaccharide with potential applications in chemotherapy and as a sweetener (Hofmann et al. 2005). l-Mannose has much potential applications in food and pharmaceutical industry as an unnatural sugar (Bhuiyan et al. 1997). Thus, the discovery of a thermostable RhaA from T. maritima that can be used to generate these l-form monosaccharides is of major industrial and medicinal importance.
In summary, a gene encoding the putative protein from T. maritima was cloned, expressed, purified, and characterized. Evaluation of the substrate specificity of the expressed enzyme identified it as a RhaA. Since RhaA from T. maritima exhibited high activity for l-form monosaccharides such as l-rhamnose, l-lyxose, l-mannose, l-xylulose, and l-fructose, the highest ever reported concentrations and productivities of l-lyxose and l-mannose were obtained. The enzyme is the first reported thermostable RhaA. Thus, the thermostable enzyme should be of great value in industrial applications.
References
Badia J, Gimenez R, Baldoma L, Barnes E, Fessner WD, Agilar J (1991) l-Lyxose metabolism employs the l-rhamnose pathway in mutant cells of Escherichia coli adapted to grow on l-lyxose. J Bacteriol 173:5144–5150
Bhuiyan SH, Itami Y, Izumori K (1997) Immobilization of l-rhamnose isomerase and its application in l-mannose production from l-fructose. J Ferment Bioeng 84:558–562
Bhuiyan SH, Itami Y, Takada G, Izumori K (1999) Preparation of l-talose and d-gulose from l-tagatose and d-sorbose, respectively, using immobilized l-rhamnose isomerase. J Biosci Bioeng 88:567–570
Bruce LZ, Henrik KN, Robert LS (1991) Thermostable enzymes for industrial applications. J Ind Microbiol Biotechnol 8:71–81
Conners SB, Montero CI, Comfort DA, Shockley KR, Johnson MR, Chhabra SR, Kelly RM (2005) An expression-driven approach to the prediction of carbohydrate transport and utilization regulons in the hyperthermophilic bacterium Thermotoga maritima. J Bacteriol 187:7267–7282
Domagk GF, Zech R (1963) On the decomposition of desoxy sugars by bacterial enzymes. I. l-Rhamnose isomerase from Lactobacillus plantarum. Biochem Z 339:145–153
Doong SL, Tsai CH, Schinazi RF, Liotta DC, Cheng YC (1991) Inhibition of the replication of hepatitis B virus in vitro by 2′,3′-dideoxy-3′-thiacytidine and related analogues. Proc Natl Acad Sci USA 88:8495–8499
Granstrom TB, Takata G, Morimoto K, Leisola M, Izumori K (2005) l-Xylose and l-lyxose production from xylitol using Alcaligenes 701B strain and immobilized l-rhamnose isomerase enzyme. Enzyme Microb Technol 36:976–981
Groussac E, Ortiz M, Francois J (2000) Improved protocols for quantitative determination of metabolites from biological samples using high performance ionic-exchange chromatography with conductimetric and pulsed amperometric detection. Enzyme Microb Technol 26:715–723
Hofmann C, Boll R, Heitmann B, Hauser G, Durr C, Frerich A, Weitnauer G, Glaser SJ, Bechthold A (2005) Genes encoding enzymes responsible for biosynthesis of l-lyxose and attachment of eurekanate during avilamycin biosynthesis. Chem Biol 12:1137–1143
Itoh H, Izumori K (1996) Enzymatic production of l-tagatose and l-fructose from l-sorbose and l-psicose, respectively. J Ferment Bioeng 81:351–353
Itoh H, Sato T, Izumori K (1995) Preparation of d-psicose from d-fructose by immobilized d-tagatose 3-epimerase. J Ferment Bioeng 80:101–103
Izumori K, Mitchell M, Elbein AD (1976) Evidence that the isomerization of d-ribose and l-rhamnose is catalyzed by the same enzyme in Mycobacterium smegmatis. J Bacteriol 126:553–555
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30
Leang K, Maekawa K, Menavuvu BT, Morimoto K, Granstrom TB, Takada G, Izumori K (2004a) A novel enzymatic approach to the massproduction of l-galactose from l-sorbose. J Biosci Bioeng 97:383–388
Leang K, Takada G, Fukai Y, Morimoto K, Granström TB, Izumori K (2004b) Novel reactions of l-rhamnose isomerase from Pseudomonas stutzeri and its relation with d-xylose isomerase via substrate specificity. Biochim Biophys Acta 1674:68–77
Leang K, Takada G, Ishimura A, Okita M, Izumori K (2004c) Cloning, nucleotide sequence, and overexpression of the l-rhamnose isomerase gene from Pseudomonas stutzeri in Escherichia coli. Appl Environ Microbiol 70:3298–3304
Moralejo P, Egan SM, Hidalgo E, Aguilar J (1993) Sequencing and characterization of a gene cluster encoding the enzymes for l-rhamnose metabolism in Escherichia coli. J Bacteriol 175:5585–5594
Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Nelson WC, Ketchum KA, McDonald L, Utterback TR, Malek JA, Linher KD, Garrett MM, Stewart AM, Cotton MD, Pratt MS, Phillips CA, Richardson D, Heidelberg J, Sutton GG, Fleischmann RD, Eisen JA, White O, Salzberg SL, Smith HO, Venter JC, Fraser CM (1999) Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature 399:323–329
Oudega B, Koningstein G, Rodrigues L, de Sales Ramon M, Hilbert H, Dusterhoft A, Pohl TM, Weitzenegger T (1997) Analysis of the Bacillus subtilis genome: cloning and nucleotide sequence of a 62 kb region between 275 degrees (rrnB) and 284 degrees (pai). Microbiology 143:2769–2774
Poonperm W, Takata G, Morimoto K, Granstrom TB, Izumori K (2007a) Production of l-xylulose from xylitol by a newly isolated strain of Bacillus pallidus Y25 and characterization of its relevant enzyme xylitol dehydrogenase. Enzyme Microb Technol 40:1206–1212
Poonperm W, Takata G, Okada H, Morimoto K, Granstrom TB, Izumori K (2007b) Cloning, sequencing, overexpression and characterization of l-rhamnose isomerase from Bacillus pallidus Y25 for rare sugar production. Appl Microbiol Biotechnol 76:1297–1307
Rao D, Gullapalli P, Yoshihara A, Jenkinson SF, Morimoto K, Takata G, Akimitsu K, Tajima S, Fleet GW, Izumori K (2008) Direct production of l-tagatose from l-psicose by Enterobacter aerogenes 230S. J Biosci Bioeng 106:473–480
Acknowledgments
This work was supported by the Korea Science and Engineering Foundation (KOSEF) through the National Research Lab. Program funded by the Ministry of Education, Science and Technology (R0A-2007-000-20015-0) and by a grant (Code #2007-0301034024) from BioGreen 21 Program, Rural Development Administration.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Park, CS., Yeom, SJ., Lim, YR. et al. Characterization of a recombinant thermostable l-rhamnose isomerase from Thermotoga maritima ATCC 43589 and its application in the production of l-lyxose and l-mannose. Biotechnol Lett 32, 1947–1953 (2010). https://doi.org/10.1007/s10529-010-0385-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-010-0385-7