
Abstract
Expression of proteins on the surface of yeasts has a wide range of applications in biotechnology, such as directed evolution of proteins for increased affinity and thermal stability, screening of antibody libraries, epitope mapping, and use as whole-cell biocatalysts. However, hyperglycosylation can interfere with overall protein accessibility on the surface. Therefore, the less elaborate hyperglycosylation in wild type Pichia pastoris and the availability of glycoengineered strains make this yeast an excellent alternative for surface display of glycoproteins. Here, we report the implementation of the well-established a-agglutinin-based yeast surface display technology in P. pastoris. Four heterologous proteins were expressed on the surface of a wild type and a glycoengineered strain. Surface display levels were monitored by Western blot, immunofluorescence microscopy, and FACS analysis. The availability of glycoengineered strains makes P. pastoris an excellent alternative for surface display of glycoproteins and paves the way for new applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Surface expression of proteins has a wide range of applications in biotechnology, e.g. directed evolution of proteins, screening of non-immune antibody libraries, epitope mapping, and development of whole-cell biocatalysts (Gai and Wittrup 2007).
Many secreted proteins are post-translationally modified in the endoplasmic reticulum (ER), e.g. by N-glycosylation, which is particularly important because it often assists in the folding of glycoproteins (Helenius and Aebi 2004). However, it has been reported that extensive N-hyperglycosylation can interfere with overall protein accessibility on the surface of Saccharomyces cerevisiae (Ryckaert et al. 2005). The key hyperglycosylation enzyme is Och1p, an α-1,6-mannosyltransferase that initiates hypermannosylation (Nakayama et al. 1992). Hence, OCH1 disruption reduces the extent of N-glycosylation, and thereby potentially increases the accessibility of the surface-expressed proteins (Ryckaert et al. 2005). However, glycoengineered S. cerevisiae strains are crippled. They are temperature sensitive, prone to stress, and have a clumpy morphology and doubled generation time, and so they are very difficult to handle (Nakanishi-Shindo et al. 1993). Moreover, strains with a fully humanized N-glycosylation pathway are not available.
Contrary to that, hyperglycosylation in wild type Pichia pastoris strains is less elaborate and this is probably why inactivation of Pichia OCH1 does not severely impede growth (Vervecken et al. 2004). Hence, a surface-expression system based on glycoengineered P. pastoris (Pichia surface display: PSD) could present a major improvement for the surface expression of many (glyco)proteins. Furthermore, the availability of glycoengineered Pichia strains with fully human N-glycosylation (Vervecken et al. 2004, Hamilton et al. 2006) paves the way for new applications.
Recently, several groups described the use of wild type P. pastoris for surface-display of heterologous proteins. Two groups have used the S. cerevisiae α-agglutinin to anchor their protein of interest at the Pichia cell surface (Mergler et al. 2004, Wang et al. 2007). Three other groups have used S. cerevisiae Flo1p as an anchor protein (Tanino et al. 2006, Jiang et al. 2007, Ren et al. 2007). To our knowledge, however, no reports have been published describing the implementation of the commercially available a-agglutinin-based yeast surface display system (Invitrogen) in P. pastoris.
Here we report a novel a-agglutinin-based system for display of heterologous proteins on the cell surface of P. pastoris and its validation in both wild type and glycoengineered strains.
Methods
Strains and media
Escherichia coli MC1061 was used as the host strain for recombinant DNA manipulations. Pichia pastoris GS115 (Invitrogen) and Man5 strains (Vervecken et al. 2004) were used for the surface-expression of heterologous proteins. E. coli was cultivated in LB medium (10 g Tryptone/l, 5 g yeast extract/l, and 5 g NaCl/l) containing 100 μg ampicillin/ml or 25 μg zeocin/ml. P. pastoris was grown in BMGY medium [10 g yeast extract/l, 20 g peptone/l, 13.4 g yeast nitrogen base/l, 100 mM potassium phosphate buffer (pH 6.0), and 10 g glycerol/l] and heterologous protein expression was induced in BMMY medium [10 g yeast extract/l, 20 g peptone/l, 13.4 g yeast nitrogen base/l, 100 mM potassium phosphate buffer (pH 6.0), and 10 ml methanol/l].
Construction of expression plasmids
The AGA1 ORF (encoding the a-agglutinin anchor subunit Aga1p) was PCR amplified from Saccharomyces cerevisiae genomic DNA and ligated to the NotI/EcoRI-opened P. pastoris vector pPICZA (Invitrogen), resulting in pPICZAAGA1. The AGA2 expression cassette was amplified by PCR using the S. cerevisiae vector pYD1.1 as a template. The resultant amplicon was cloned EcoRI/BamHI in the P. pastoris vector pPIC9 (Invitrogen), yielding pPIC9AGA2. Plasmid pYD1.1 was constructed by digesting the commercially available pYD1 vector (Invitrogen) with EcoRI and BamHI, blunting with T4 DNA polymerase, and religating.
Four heterologous genes were cloned in pPIC9AGA2: human galectin-1, human erythropoietin, mouse interferon gamma and mouse interferon beta, resulting in pPIC9AGA2hGal, pPIC9AGA2hEPO, pPIC9AGA2mIFNγ and pPIC9AGA2mIFNβ, respectively. All four ORFs were PCR-amplified with gene-specific primers (Supplementary Table 1) from plasmids carrying the ORFs and XhoI/ApaI-cloned in pPIC9AGA2. The human galectin-1 ORF was PCR-amplified using PSDhGal1S and PSDhGal1AS as primers. The human erythropoietin ORF was PCR amplified using PSDhEPOS and PSDhEPOAS. The mouse interferon gamma ORF was PCR amplified using PSDmIFNgS and PSDmIFNgAS. The mouse interferon beta ORF was PCR amplified using PSDmIFNbS and PSDmIFNbAS, and cloned in the XhoI(blunted)/ApaI-opened pPIC9AGA2 vector.
Pichia transformation
Linearized vectors were transformed into P. pastoris as described (Cregg and Russel 1998). Genomic integration was confirmed by performing PCR on genomic DNA.
Cultivation conditions
All P. pastoris cells were grown to saturation in 10 ml BMGY medium in 50 ml Falcon tubes at 30°C. After 48 h of cultivation, the cultures were centrifuged at 3,000g for 5 min and resuspended in BMMY medium. The induction was continued for another 48 h by adding 100 μl of 100% methanol to the cultures twice daily.
Immunofluorescence microscopy and FACS analysis
Induced cells were washed twice in ice-cold water and resuspended at 4°C in PBS (pH 7.2) supplemented with 0.5 mg BSA/ml. The cells from 1 ml culture at OD600 = 1 (about 2 × 107 c.f.u.) were incubated with a 1:1,000 dilution of antibody (anti-Xpress or anti-V5) (Invitrogen) for 30 min at 4°C. After washing twice with PBS (pH 7.2) supplemented with 0.5 mg BSA/ml, the cells were incubated in the same buffer with a 1:1,000 dilution of a secondary goat anti-mouse antibody conjugated with Alexa fluor 488 (Molecular Probes, Eugene, OR) for 20 min at 4°C. After two more washing steps the cells were examined with an Axiophot fluorescence microscope (Zeiss) or with a FACSCalibur fluorescence activated cell sorter (Becton Dickinson).
The percentage of positive cells is indicated in the FACS histograms for each strain-protein combination. This number corresponds to the percentage of cells that fell outside a FL1/FSC gate encompassing >99% of the negative control cells. The increase in fluorescence relative to autofluorescence of the negative control was also calculated for each strain-protein combination.
Western blot analysis
Approximately 108 induced cells (277 μl from culture at OD600 = 18) were washed twice with ice-cold water, resuspended in PBS supplemented with 10 mM DTT, and incubated for 3 h at room temperature to reduce the disulfide bonds between Aga2p and Aga1p. They were pelleted by centrifugation and the supernatant was precipitated with 10% (w/v) trichloroacetic acid (TCA) and 0.5 mg desoxycholate/ml. Proteins were separated by 15% SDS-PAGE and transferred to a Hybond-C membrane (Amersham). Aga2-mIFN-γ was detected with anti-V5 monoclonal antibody (1:5,000 dilution; Invitrogen). The secondary antibody was horseradish peroxidase-conjugated goat anti-mouse IgG (1:10,000), and detection was performed with chemiluminescence (Western Lightning Chemiluminescence Reagent Plus kit, Perkin-Elmer).
Results and discussion
Vector control experiments
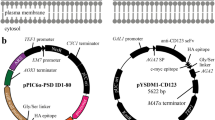
The commercially available yeast surface display (YSD) system (Invitrogen) uses the heterodimeric S. cerevisiae a-agglutinin adhesion receptor to display proteins on the cell surface of S. cerevisiae (Fig. 1a). To adopt this system in P. pastoris, we constructed two vectors. The first, pPICZAAGA1, contains the AGA1 gene under transcriptional control of the methanol-inducible AOX1 promoter (Fig. 1b). Transformation of this vector into the P. pastoris GS115 and Man5 strains resulted in GS115A1 and Man5A1, respectively. Man5 is a glycoengineered strain derived from the GS115 wild type strain (Vervecken et al. 2004) and modifies its glycoproteins predominantly with Man5GlcNAc2 N-glycans. The second vector, pPIC9AGA2, is derived from the commercially available YSD vector pYD1 (Invitrogen) and contains the AGA2 gene under AOX1 transcriptional control (Fig. 1c). Cloning the gene of interest (termed X) in frame with AGA2 produces pPIC9AGA2X. Upon transformation of pPIC9AGA2X into GS115A1 and Man5A1 two new strains are generated: GS115A1A2X and Man5A1A2X, respectively. Both strains display protein X on their surface upon methanol induction. The Aga2p-Xp fusion protein contains three epitope tags for detection: an Xpress tag between Aga2p and Xp, and a V5 and a 6xHis tag at the C-terminus of Xp (Fig. 1a).

(a) Yeast surface display. The coding sequence of the protein to be displayed (termed “X”) is cloned in frame with the C-terminal end of AGA2. Three tags (Xpress, V5 and 6xHIS) facilitate detection of the a-agglutinin-Xp fusion protein; (b) pPICZAAGA1; (c) pPIC9AGA2
As proof of principle, we transformed the “empty” pPIC9AGA2 vector (i.e., no heterologous gene inserted) into the GS115A1 and Man5A1 strains. a-Agglutinin surface expression was detected by indirect immunofluorescent labeling of the Xpress epitope tag and flow cytometry. Over 95% of the cells expressed a-agglutinin on their surface, in both the wild type and the Man5 strain (Fig. 2). This indicates that altering the N-glycosylation pathway of P. pastoris has very little influence on its surface display capabilities. However, a small difference between the two strains became apparent, especially when inducing for <24 h: on average, the wild type strain showed 2 to 3-fold more fluorescence than the Man5 strain. These data demonstrate that the S. cerevisiae a-agglutinin can be expressed on the surface of P. pastoris. However, as observed for secreted protein yields, the extent of surface display was influenced by general growth conditions, such as the duration of the methanol induction phase.

FACS analysis of GS115A1A2 and Man5A1A2. The a-agglutinin-expressing GS115 (GS115A1A2) and Man5 strains (Man5A1A2) were labeled with an anti-Xpress antibody. The increase in fluorescence is about 2.5 times higher for the GS115A1A2 strain, compared to the Man5A1A2 strain. Unlabeled cells (negative control) are shown as unfilled histograms
Surface expression of four mammalian proteins
To further evaluate the PSD technology, we expressed four mammalian proteins on the surface of P. pastoris: human erythropoietin (hEPO), mouse interferon beta (mIFN-β), mouse interferon gamma (mIFN-γ), and human galectin-1 (hGal-1). We focused on cytokines and cytokine-like proteins that are, according to the literature and to our unpublished results, difficult to produce as secreted proteins in P. pastoris. Human galectin-1 was included because it can be expressed at high levels on the surface of S. cerevisiae (Ryckaert et al. 2008).
Four vectors were constructed. pPIC9AGA2hEPO, pPIC9AGA2mIFNβ and pPIC9AGA2mIFNγ were transformed into both the GS115A1 and Man5A1 strains, whereas pPIC9AGA2hGal was transformed only into the Man5A1 strain. For each vector-strain combination, 10–20 clones were analyzed genomically by PCR (data not shown). Subsequently, 5–10 positive clones were analyzed by FACS screening. The results are shown in Fig. 3. For all the proteins we tested, the Man5 strains generated somewhat stronger fluorescent signals during FACS analysis than the GS115 strains. One explanation is the better accessibility of fusion proteins on the Man5 strains due to absence of hyperglycosyl structures on the surface-expressed heterologous proteins and on the mannoproteins that constitute the outer layer of the cell wall. Alternatively, the heterologous proteins may be more abundant on the Man5 strain, but this is less likely because expression studies of several heterologous glycoproteins in the GS115 and the Man5 strains showed no differences in protein yield (our unpublished results).

FACS analysis of surface-expressed mammalian proteins. Expression levels of the four mammalian test proteins on the surfaces of GS115 and Man5 cells as determined by flow cytometry. Human galectin-1 was most abundant on the Man5 strain, with about 95% of the cells expressing the protein on their cell wall and exhibiting 16-fold more fluorescence than the negative control. Mouse IFN-β was the least well expressed; only about 5% of Man5 cells and less than 1% of GS115 cells were positive. Surface expression levels of mIFN-γ (~55% positive cells in both strains and a 5 to 7-fold increase in fluorescence) and hEPO (7% positive in GS115 and 15% positive in Man5, corresponding to 2.5 and 4-fold increase in fluorescence, respectively) were intermediate between those of hGal-1 and mIFN-β. Unlabeled cells (negative control) are shown as unfilled histograms
Immunofluorescence microscopy and western blot analysis
Figure 4a shows the light and immunofluorescence micrographs of Man5A1A2mIFNγ cells. Daughter cells express more IFN-γ on their surface than mother cells do. This phenomenon has been reported for S. cerevisiae as well and was suggested to be due to the polarized growth morphology of yeast (Feldhaus et al. 2003). Cell wall biosynthesis in daughter cells probably results in greater incorporation of a-agglutinin in the cell wall.

Immunofluorescence and Western blot analysis. Immunofluorescence micrograph (a) and light micrograph (b) of Man5A1A2mIFNγ cells. Daughter cells express more mIFN-γ on their surface than mother cells do; (c) Western blot analysis of the GS115A1A2mIFNγ and the Man5A1A2mIFNγ strains. After induction, the proteins in the supernatant of 108 cultured cells were TCA precipitated (medium fraction), while the cultured cells were treated with DTT to reduce to disulfide bonds between Aga1p and Aga2p. The proteins detached from 108 cells were TCA-precipitated (DTT fraction). The GS115A1 and Man5A1 strains were used as a negative control. The Aga2p-mIFNγ fusion protein, with a predicted MW of 33 kDa, was detected in the DTT-extracted fractions of both strains. However, some additional lower molecular weight bands were present in the DTT-extracted fractions of the GS115A1A2mIFNγ strain, probably caused by proteolysis. In the culture medium of the Man5A1A2mIFNγ strain, an Aga2-mIFN-γ specific band of the expected size was observed as well
Finally, the presence of mIFN-γ on the Pichia cell surface was determined by Western blot analysis. The results are shown in Fig. 4b. The Aga2-mIFN-γ fusion protein was detected in the DTT-extracted fractions of the GS115A1A2mIFNγ and Man5A1A2mIFNγ strains. Upon longer incubation times, the additional lower molecular weight bands present in the DTT-extracted fraction of the GS115A1A2mIFNγ strain could be revealed in the DTT-fraction of the Man5A1A2mγ as well (data not shown). The Aga2-mIFN-γ specific band in the culture medium of the Man5A1A2mIFNγ strain could be detected in the GS115A1A2mIFNγ culture supernatant as well when longer exposure times were used (data not shown). Since a-agglutinin is covalently linked to the β-glucan in the cell wall, the altered composition of the cell wall of the glycoengineered Man5 strain might result in partial secretion of the a-agglutinin-mIFNγ fusion protein into the culture medium. The hyperglycosyl N-glycans are a major constituent of the outer layer of the yeast cell wall and serve mainly as a permeability barrier (Klis et al. 2002). In the Man5 strain, however, these hyperglycosyl structures have been trimmed to Man5GlcNAc2 oligosaccharides, thereby increasing the permeability of the outer cell wall layer and potentially decreasing the time available for cell wall proteins, such as a-agglutinin, to attach to the β-glucan.
V5 versus Xpress
We used the C-terminal V5 tag for detection in all FACS experiments involving any of the four surface-expressed heterologous proteins, because a positive signal ensures that the entire ORF is present. The same holds true for the 6xHis tag. A positive Xpress signal, however, demonstrates the presence of Aga2p on the surface, but the surface-expressed protein might have been partially or completely degraded by intra- or extracellular proteases. Hence, analysis based on the Xpress tag could be misleading. This is illustrated in Fig. 5 for Trypanosoma cruzi trans-sialidase expressed on the surface of the Man5 strain. These data show that proteases released into the medium by P. pastoris can affect the integrity of the surface-expressed Aga2p fusion protein in such a way that the C-terminal tag (V5 or E-tag) is lost while the Xpress tag is maintained. However, it should be noted that addition of protease inhibitors did not improve the V5-signal for all proteins tested. This shows, as has been reported (Shusta et al. 1999), that intracellular proteases might also be responsible for partial degradation of the Aga2p-fusion protein, such that Aga2p appears on the surface without a fusion partner or with a truncated one. This easy test yields information on the protease stability of the displayed protein, and therefore indicates whether adding protease inhibitors to the culture medium could increase yields of a given protein during expression.

Trans-sialidase surface expression—V5 versus Xpress. Surface expression levels of T. cruzi trans-sialidase in the Man5 strain in the absence (a) and presence (b) of protease inhibitors. Cells were labeled with an anti-Xpress or an anti-E-tag antibody. Addition of protease inhibitors during induction clearly results in improved E-tag signals, while the Xpress signal remains the same
PSD versus YSD
Comparison of our results in P. pastoris with data from the literature and with our unpublished results in S. cerevisiae shows some differences between YSD and PSD.
First, only a subset of the WT surface-displaying S. cerevisiae cells scored positive in FACS analysis. This resulted in the invariable presence in histograms of two clearly distinct populations of cells, i.e. expressers and non-expressers. The existence of a non-expressing population can be ascribed to several causes: (1) plasmid loss; pYD1 is an episomal vector and loss of selective pressure will result in curing of the plasmid; (2) mother cells that do not express the fusion protein on their cell walls due to the polarized growth morphology of yeast; (3) cells at a stage of growth or induction that are incapable of expressing a surface fusion protein (Feldhaus et al. 2003).
However, during FACS analysis, surface-displaying P. pastoris cells appear as only one population of cells, i.e. a non-expressing population like that seen in S. cerevisiae is absent, and all cells express, but to different extents. Although both yeasts belong to the Saccharomycetaceae family, certain structural, biochemical and metabolic differences between them can explain this difference. For example, the S. cerevisiae Golgi apparatus differs considerably from the more human-like Golgi of P. pastoris (Mogelsvang et al. 2003; Losev et al. 2006), which might affect the secretion and glycosylation machineries and the cell wall composition.
A second difference between PSD and YSD lies in the characteristics of the vectors used with them. YSD in its most commonly used form relies on episomal vectors, and the high transformation efficiencies that can be achieved are beneficial, for example when constructing libraries. However, these vectors may be lost when cells are grown under non-selective conditions, and so the cells should not be grown in rich media. In contrast, PSD uses integrative vectors, resulting in stable transformants that do not require continuous selective pressure to prevent plasmid loss, and can be grown in both rich and minimal media. The current Pichia transformation procedure (Cregg and Russel 1998) allows construction of libraries of larger size (5 × 107 to 108 clones; our unpublished results).
A third major difference between PSD and YSD is the availability of glycoengineered Pichia strains with wild type growth characteristics that can modify glycoproteins with fully humanized N-glycans. Combination of PSD and glycoengineered strains with a fully humanized N-glycosylation pathway opens the door for new biotechnological and therapeutic applications.
Conclusions
In recent years, extensively glycoengineered P. pastoris strains have been generated both by our group and by researchers at GlycoFi Inc. (recently acquired by Merck). Several of these strains homogeneously modify their glycoproteins with a high-mannose, a hybrid, or a complex human-like N-glycan structure. Here we have shown that our PSD technology is compatible with at least one of these strains (Man5). We are now testing how more extensively glycoengineered strains behave when they display heterologous proteins on their cell surface. The availability of these glycoengineered strains to the scientific community might pave the way for new applications that are not feasible using the S. cerevisiae based yeast surface display system.
Abbreviations
- ER:
-
Endoplasmic reticulum
- PSD:
-
Pichia surface display
- YSD:
-
Yeast surface display
- hEPO:
-
Human erythropoietin
- hGal-1:
-
Human galectin-1
- mIFN-β:
-
Mouse interferon beta
- mIFN-γ:
-
Mouse interferon gamma
- FACS:
-
Fluorescence activated cell sorting
References
Cregg JM, Russel KA (1998) Transformation. In: Higgins DR, Cregg JM (eds) Pichia protocols, 1st edn. Humana Press, Totowa, NJ, pp 27–39
Feldhaus MJ, Siegel RW, Opresko LK et al (2003) Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat Biotechnol 21:163–170
Gai SA, Wittrup KD (2007) Yeast surface display for protein engineering and characterization. Curr Opin Struct Biol 17:467–473
Hamilton SR, Davidson RC, Sethuraman N, et al (2006) Humanization of yeast to produce complex terminally sialylated glycoproteins. Science 313:1441–1443
Helenius A, Aebi M (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem 73:1019–1049
Jiang ZB, Song HT, Gupta N et al (2007) Cell surface display of functionally active lipases from Yarrowia lipolytica in Pichia pastoris. Protein Expr Purif 56:35–39
Klis FM, Mol P, Hellingwerf K et al (2002) Dynamics of cell wall structure in Saccharomyces cerevisiae. FEMS Microbiol Rev 26:239–256
Losev E, Reinke CA, Jellen J et al (2006) Golgi maturation visualized in living yeast. Nature 441:1002–1006
Mergler M, Wolf K, Zimmermann M (2004) Development of a bisphenol A-adsorbing yeast by surface display of the Kluyveromyces yellow enzyme on Pichia pastoris. Appl Microbiol Biotechnol 63:418–421
Mogelsvang S, Gomez-Ospina N, Soderholm J et al (2003) Tomographic evidence for continuous turnover of Golgi cisternae in Pichia pastoris. Mol Biol Cell 14:2277–2291
Nakanishi-Shindo Y, Nakayama K, Tanaka A et al (1993) Structure of the N-linked oligosaccharides that show the complete loss of alpha–1, 6-polymannose outer chain from och1, och1 mnn1, and och1 mnn1 alg3 mutants of Saccharomyces cerevisiae. J Biol Chem 268:26338–26345
Nakayama K, Nagasu T, Shimma Y et al (1992) OCH1 encodes a novel membrane bound mannosyltransferase: outer chain elongation of asparagine-linked oligosaccharides. EMBO J 11:2511–2519
Ren R, Jiang Z, Liu M et al (2007) Display of adenoregulin with a novel Pichia pastoris cell surface display system. Mol Biotechnol 35:103–108
Ryckaert S, Martens V, De Vusser K et al (2005) Development of a S. cerevisiae whole cell biocatalyst for in vitro sialylation of oligosaccharides. J Biotechnol 119:379–388
Ryckaert S, Callewaert N, Jacobs PP et al (2008) Fishing for lectins from diverse sequence libraries by yeast surface display—an exploratory study. Glycobiology 18:137–144
Shusta EV, Kieke MC, Parke E et al (1999) Yeast polypeptide fusion surface display levels predict thermal stability and soluble secretion efficiency. J Mol Biol 292:949–956
Tanino T, Fukuda H, Kondo A (2006) Construction of a Pichia pastoris cell-surface display system using Flo1p anchor system. Biotechnol Prog 22:989–993
Vervecken W, Kaigorodov V, Callewaert N et al (2004) In vivo synthesis of mammalian-like, hybrid-type N-glycans in Pichia pastoris. Appl Environ Microbiol 70:2639–2646
Wang Q, Li L, Chen M et al (2007) Construction of a novel system for cell surface display of heterologous proteins on Pichia pastoris. Biotechnol Lett 29:1561–1566
Acknowledgements
Pieter Jacobs is supported by a PhD grant from the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen). This research was funded by Research Corporation Technologies (Tucson, AZ). The continuous support and encouragement of Dr. Bennet Cohen is very much appreciated. Eef Parthoens and Wies Deckers are acknowledged for their help with the immunofluorescence microscopy. We thank Dr. Amin Bredan for editing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Jacobs, P.P., Ryckaert, S., Geysens, S. et al. Pichia surface display: display of proteins on the surface of glycoengineered Pichia pastoris strains. Biotechnol Lett 30, 2173–2181 (2008). https://doi.org/10.1007/s10529-008-9807-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-008-9807-1