
Abstract
Yeast surface display (YSD) has been shown to represent a powerful tool in the field of antibody discovery and engineering as well as for selection of high producer clones. However, YSD is predominantly applied in Saccharomyces cerevisiae, whereas expression of heterologous proteins is generally favored in the non-canonical yeast Pichia pastoris (Komagataella phaffii). Establishment of surface display in P. pastoris would therefore enable antibody selection and expression in a single host. Here we describe the generation of a Pichia surface display (PSD) system based on antibody expression from episomal plasmids. By screening a diverse set of expression vectors using Design of Experiments (DoE), the effect of different genetic elements on the surface expression of antibody fragments was analyzed. Among the tested genetic elements, we found that the combination of P. pastoris formaldehyde dehydrogenase (FLD1) promoter, S. cerevisiae invertase 2 signal peptide (SUC2), and α-agglutinin cell wall protein (SAG1) including an autonomously replicating sequence of Kluyveromyces lactis (panARS) were contributing most strongly to higher display levels of three tested antibody fragments. Employing this combination resulted in the display of antibody fragments for up to 25% of cells. Despite significantly reduced expression levels in PSD compared to well-established YSD in S. cerevisiae, similar fractions of antigen binding single-chain variable fragments (scFvs) were observed (80% vs. 84%). In addition, plasmid stability assays and flow cytometric analysis demonstrated the efficient plasmid clearance of cells and associated loss of antibody fragment display after removal of selective pressure.
Key points
• First report of antibody display in P. pastoris using episomal plasmids.
• Identification of genetic elements conferring highest levels of antibody display.
• Comparable antigen binding capacity of displayed scFvs for PSD compared to YSD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The surface display of proteins is an efficient method to directly link observed phenotypic properties of proteins to the encoding genetic information. It is representing one of the most important high-throughput methodologies in biotechnology and has a wide range of applications, such as directed evolution of proteins, screening of antibody libraries, epitope mapping, and development of whole-cell biocatalysts (Gai and Wittrup 2007). Protein display was initially established in bacteriophages (McCafferty et al. 1990; Scott and Smith 1990), but could be extended to various microorganisms with advancing genetic engineering (Boder and Wittrup 1997; Ståhl and Uhlén 1997). Especially YSD in Saccharomyces cerevisiae has achieved high popularity and success (Andreu and Olmo 2018; Cherf and Cochran 2015). However, proteins expressed by S. cerevisiae tend to be hyperglycosylated, which might hamper their activity or interfere with the protein accessibility on the cell surface (Ryckaert et al. 2005; Tang et al. 2016). Surface display of proteins has been established in Pichia pastoris as well, besides other biotechnologically used yeasts (Mergler et al. 2004). In contrast to the widely used YSD, the PSD systems commonly utilize a single cell wall protein (CWP) instead of independently expressed heterodimeric S. cerevisiae a-agglutinin subunits (AGA1/AGA2), thereby simplifying the surface display. In both systems, surface targeting of heterologous proteins is facilitated by fusion to a specific anchor domain of CWPs. For PSD, several heterologous proteins, including antibodies, have been successfully displayed using different CWPs such as a protein with internal repeats (PIR1) from P. pastoris and flocculation protein (FLO1), cell wall glycoprotein (SED1) as well as α-agglutinin (SAG1) from S. cerevisiae (Mergler et al. 2004; Su et al. 2010; Tanino et al. 2007; Khasa et al. 2011). With the development of glycoengineered Pichia strains exhibiting a human-like glycan profile, this expression host is able to express glycoproteins suitable for therapeutic applications (Li et al. 2006; Liu et al. 2013; Vervecken et al. 2004). This in combination with tightly regulated promoters, versatile genetic engineering toolsets, growth on cheap carbon sources, as well as growth to high cell densities has substantiated the application of P. pastoris as an important expression system for biotechnological and biopharmaceutical proteins (Karbalaei et al. 2020; Kim et al. 2015).
PSD has almost exclusively been focused on the display of single proteins and screening of complex protein libraries has been limited to a few studies in the past. Ryckaert et al. (2010) have displayed camelid heavy chain nanobodies fused to the N-terminus of S. cerevisiae SAG1 on P. pastoris in order to screen an immune library of single domain antibodies. Other projects analyzed the display of antibody fragments or full-length IgG antibodies on the surface of P. pastoris, which requires post-translational assembly of the heavy chain and the light chain, and demonstrated their functional display by selecting antibodies with higher affinities (Lin et al. 2012; Shaheen et al. 2013).
Despite the fact that expression of heterologous proteins in P. pastoris is usually facilitated by vector integration, several studies have demonstrated successful expression based on episomal plasmids (Lee et al. 2005; Sasagawa et al. 2011). In some cases, these were even outperforming genomically integrated controls by several fold (Camattari et al. 2016; Obst et al. 2017). Especially in the context of protein display and engineering, the use of episomal plasmid display systems would be more favorable since higher transformation efficiencies can be achieved when using circular, P. pastoris-specific ARS (PARS1) containing plasmids (Cregg et al. 1985). Those elements confer initiation of replication independent from the genome and influence the stability, transformation efficiency, and copy number of an episomal plasmid (Peña et al. 2018). With the development of CRISPR-Cas genome editing tools for P. pastoris (Weninger et al. 2016), PARS1 has been employed due to efficient plasmid clearance upon removal of selective pressure. Several recently identified autonomously replicating sequences (ARS) enable maintenance of episomal plasmids and efficient expression of recombinant proteins (Liachko and Dunham 2014; Nakamura et al. 2018; Schwarzhans et al. 2017), but were also tested for the development and optimization of the CRISPR-Cas9-based genome editing system, since they are inherently unstable in P. pastoris (Gu et al. 2019).
In this study, a Design of Experiments screening was employed in order to analyze the designed episomal vector set for a preferential combination of genetic elements known to potentially influence protein expression levels, such as promoters, signal peptides, cell wall proteins, and ARS. Based on the identified elements conferring highest surface expression of antibody fragments, we have developed a novel antibody surface display system in P. pastoris. We subsequently examined display efficiency and functional display of antibody fragments by analyzing expression levels and antigen binding capacity in comparison to the well-established YSD in S. cerevisiae. The surface capture of proteins was not limited to a clinically relevant antigen, the cluster of differentiation 123 (CD123), but could also be demonstrated for antibody-based products used in the biopharmaceutical industry, such as antigen-binding fragments (Fab). Lastly, we have demonstrated the switchable/transient nature of scFv display in our PSD system, which might pave the way for a new selection method of producer clones that can be directly used for subsequent production purposes.
Materials and methods
Strains and media
Escherichia coli NEB® 10-beta (New England Biolabs, Ipswich, MA, USA) was cultivated in LB medium (10 g/l tryptone, 5 g/l yeast extract, and 5 g/l NaCl) containing either 50 µg/ml blasticidin or 50 µg/ml kanamycin or 50 µg/ml nourseothricin for plasmid amplification. Saccharomyces cerevisiae EBY100 (ATCC® MYA-4941, Manassas, VA, USA) and P. pastoris (Komagataella phaffii) CBS7435 mutS (ΔAOX1::FRT) (Näätsaari et al. 2012, Graz Pichia Pool) were used for surface expression of heterologous proteins. This mutant strain was employed as the strong production of alcohol oxidase (AOX1) in WT (mut+) strains during growth on methanol may compete with the production of recombinant proteins. For yeast cultivation in rich media, cells were grown in YPD medium (10 g/l yeast extract, 20 g/l peptone, and 20 g/l glucose). For selective growth and induction of protein expression under selective conditions, S. cerevisiae was cultivated in SDCAA (20 g/l glucose, 6.7 g/l yeast nitrogen base, 5 g/l Bacto casamino acids, 5.4 g/l Na2HPO4, 8.56 g/l NaH2PO4) and SGCAA medium (same as SDCAA, but with 20 g/l galactose instead of glucose), respectively, as described by Chao et al. (2006). P. pastoris was grown in BMD (Buffered Minimal Dextrose) (10 g/l glucose, 13.4 g/l yeast nitrogen base, 0.5 µg/ml biotin, 200 mM potassium phosphate buffer pH 6.0) containing 100–500 µg/ml nourseothricin. Heterologous protein expression was induced in BMD/BMG (Buffered Minimal Glycerol)/BMM (Buffered Minimal Methanol) medium (13.4 g/l yeast nitrogen base, 0.5 µg/ml biotin, 200 mM potassium phosphate buffer pH 6.0 and either 10 g/l glucose, 10 g/l glycerol, or 5 g/l methanol) supplemented with 500 µg/ml nourseothricin, respectively.
Construction and transformation of expression plasmids
In order to facilitate a high-throughput cloning strategy, the P. pastoris vector pPIC6α A (Thermo Fisher Scientific Inc., Waltham, MA, USA) was modified for Golden Gate assembly (Engler et al. 2008) by introducing type IIs recognition sites (BsaI) upstream of the promoter and downstream of the AOX1 terminator region. The two existing BsaI recognition sites in the vector backbone were eliminated by site directed mutagenesis using primers pDGN07-10 (Table S1). To enable blue/white selection for screening of positive E. coli clones, the β-galactosidase α-fragment coding sequence (lacZ’, GenBank Accession No. AAB18068.1) including the lac promoter, flanked by BsaI recognition sites, that would result in CCAT and AGCT fusion sites, were introduced into the vector. This synthesized sequence (ATUM, Newark, CA, USA) was cloned in the vector pPIC6α A using BglII/BamHI restriction sites, yielding pPIC6α-lacZ. Additionally, the resistance marker of the vector was exchanged for the NrsR marker. The NAT1 coding sequence of Streptomyces noursei (GenBank Accession No. X73149.1) was synthesized (Integrated DNA Technologies Inc., Coralville, IA, USA) with NcoI and EcoRV restriction sites and ligated to the NcoI/EcoRV hydrolyzed vector pPIC6α-lacZ, resulting in the destination vector pPIC6α-GG for Golden Gate cloning. Elements of the basic module libraries (endogenous P. pastoris promoters: alcohol dehydrogenase (ADH1), alternative oxidase (AOD), AOX1, enolase (ENO1), and FLD1; signal peptides: S. cerevisiae α-mating factor (α-MF), α-MF:Δ57-70 and SUC2; antibody fragments: anti-human CD123 scFv, anti-human IgG Fab region scFv and anti-6 × histidine single variable domain (VHH) antibody; CWPs: SAG1, SED1, and PIR1; ARSs: panARS, P. pastoris mitochondrial DNA (mtDNA), and PARS1; see Fig. 1c) were synthesized by ATUM and in case for the non-yeast-derived sequences (all antibody fragment sequences), they were codon optimized for expression in P. pastoris (for sequence overview see Table S2). Where necessary, BsaI restriction sites were removed to ensure compatibility with the assembly method. For the fusion site located upstream of the start codon, AAAC as part of the consensus Kozak sequence in P. pastoris was chosen (Vogl and Glieder 2013). The three remaining fusion sites (CCAT, TAGC, and AGCT) were all positioned in non-translated sequences. For assembly, all DNA fragments were provided as uncut plasmids and combined automatically with the vector pPIC6α-GG using an automated liquid handling system (Tecan Group Ltd., Männedorf, Switzerland). After transformation of plasmids into E. coli, white colonies from each transformation were analyzed by colony PCR. Plasmid DNA was subsequently extracted from positive clones and correct vector assembly was confirmed by DNA sequencing as well as analytical endonuclease cleavage (primer sequences for colony PCR and sequencing are listed in Table S1). Genetic elements of the display vector for S. cerevisiae pYSDM1 were derived from the from pYD1 plasmid backbone (Thermo Fisher Scientific Inc., Waltham, MA, USA). pYD1 is a display vector with the surface expression cassette located N-terminal to the Aga2p yeast membrane associated protein and under transcriptional control of the GAL1 promoter. Additional features of the pYD1 vector include a TRP1 gene for selection and a CEN6/ARS4 for stable, episomal replication in S. cerevisiae as well as an antibiotic resistance gene and the pUC origin for selection and replication in E. coli. An anti-human CD123 scFv ORF obtained from an in-house phage display library screening was NcoI/NotI-cloned in frame with the AGA2 gene, resulting in pYSDM1-CD123, and transformed into S. cerevisiae EBY100 which contains the plasmid, pIU211 stably integrated into the genome (Boder and Wittrup 1997). In both display systems, the C-terminal domain of all antibody fragments was connected to the N-terminal domain of each CWP through a flexible linker (Gly4Ser)3. Vectors pYSDM1 and pPIC6α A without gene inserts were used as controls. Yeast were transformed using the Frozen-EZ Yeast Transformation II™ Kit (Zymo Research Corporation, Irvine, CA, USA). Cultivation conditions after the transformation, screenings, and re-screenings of the generated set of expression vectors for indicated PSD transformants were performed as outlined previously (Weis et al. 2004; Obst et al. 2017). Cells were inoculated in BMD media in deep-well plates. For induction of protein expression, cells were centrifuged after ~ 60 h of cultivation and resuspended in either 5 g/l BMM used for methanol inducible constructs (PAOX1, PFLD1), 10 g/l BMD for constitutively induced constructs (PAOD), or 10 g/l BMG for glycerol inducible constructs (PADH1, PENO1), respectively, and cultivated for further 48 h. For evaluating the antigen binding capability, surface expression of single P. pastoris transformants was induced as described by Ryckaert et al. (2010). In brief, P. pastoris cells were grown for 48 h in BMG at 30 °C. To induce protein expression, cells were harvested and washed twice with PBS (pH 7.2, 1% BSA), followed by resuspension in BMM and further cultivation for 24 h at 20 °C. S. cerevisiae transformants were induced as previously described (Chao et al. 2006).

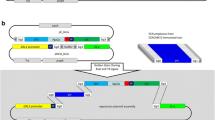
Display systems in yeast using episomal plasmids. a Schematic drawing of Pichia surface display (left) and yeast surface display in S. cerevisiae (right). The antibody fragment that should be displayed is tethered to the cell by C-terminal fusion to a cell wall protein (CWP). In P. pastoris, two GPI (SED1 and SAG1) and PpPIR1 CWPs were employed, whereas display in S. cerevisiae was facilitated by commonly used AGA1-AGA2. Two peptide tags (c-myc and HA) allow for detection of the antibody fragment-cell wall fusion protein. b Generic vector for the set of antibody display vectors used in P. pastoris, pPIC6α-PSD1-80 (left) and for the anti-human CD123 scFv display in S. cerevisiae, pYSDM1-CD123 (right). A signal peptide (SP) for protein secretion is located upstream of the to be displayed fusion protein, which is comprised of an antibody fragment followed by peptide tags, a flexible linker and a CWP. Autonomously replicating sequences (ARS) are used for episomal maintenance of display vectors in P. pastoris and S. cerevisiae. c Combinatorial design overview for assembling PSD expression vectors. P. pastoris-specific genetic elements that are known to influence protein expression levels were flanked by compatible overhangs and are interchangeable
Experimental design and analysis
All DoE studies were designed using the software JMP® version 14 (SAS Institute Inc., Cary, NC, USA). Maximizing the display efficiency of surface expressed antibody fragments was evaluated by determining cell frequency of surface expression, and median fluorescence intensity (MFI) of two fluorescence signals obtained by labeling the hemagglutinin (HA) and c-myc tag. To investigate the influence of the genetic elements, a custom screening design with 5 multilevel categorical factors was generated: promoter, signal peptide, antibody fragment, cell wall protein, and ARS (Table 1). Fraction of design space, power analysis, and color map of correlation as methods for design evaluation were chosen to evaluate the adequacy of the DoE design. Eighty P. pastoris clones harboring individual PSD constructs (see Table S3 for a complete overview designed constructs) were immunologically labeled with fluorophore conjugated antibodies specific for both peptide tags in order to detect displayed proteins and analyzed by flow cytometry. The surface expression of antibody fragments was analyzed by determining the frequency of antibody displaying cells and stain indices for APC and FITC fluorescence. After data acquisition, the collected data for each output was fitted using multiple linear regression (MLR) modelling and checked for model quality as described recently (Brown et al. 2018; Montgomery 2013). Data points exhibiting studentized residuals above 2.575 (in absolute values) were excluded from the model (outliers). Terms displaying a p-value of > 0.05 were excluded from the model according to the backward stepwise regression method. Significance of the model was determined by ANOVA. A prediction profiler was used to display the overall desirability (maximized surface expression) for each term.
Immunofluorescence microscopy and flow cytometric analysis
After induction of protein expression, approximately 1 × 106 cells were incubated with 10 µl of antibody (c-myc Antibody, anti-human/mouse/rat, mouse IgG1, FITC and/or HA antibody, mouse IgG1, APC) (130–116-485 and 130–123-553, Miltenyi Biotec, Bergisch Gladbach, Germany) for 10 min at 4 °C to label surface expressed fusion proteins. After washing with PBS (1% BSA), cells were examined with a confocal laser scanning microscope LSM 710 (Carl Zeiss AG, Oberkochen, Germany) using a BP 620/60 filter and excitation at 633 nm, or with a MACSQuant® X flow cytometer (Miltenyi Biotec). Stain indices were calculated as follows:
The amount of displayed proteins per cell was estimated for the P. pastoris strain CBS7435CD123 using Quantibrite™ PE beads (BD Bioscience, Franklin Lakes, NJ, USA). MFI was determined by labeling cells with an anti-HA antibody PE conjugate. To analyze antigen display, cells were incubated with recombinant antigens, either 100 nM CD123 (biotinylated human IL-3 R / CD123 protein, histidine, avitag™, ACROBiosystems, Newark, DE, USA), or 1 µM chimeric Fab (anti-human CD19 and CD33 chimeric mouse/human Fab, Miltenyi Biotec), for 30 min at RT and washed twice. CD123 displaying cells were labeled with an anti-biotin antibody VioBlue conjugate (Miltenyi Biotec) in addition to the anti-HA antibody APC conjugate. For labeling of chimeric Fab displaying cells, a biotinylated monoclonal antibody against poly-histidine tag was used as the primary antibody, and an anti-biotin antibody VioBlue conjugate as the secondary antibody (130–113-295, Miltenyi Biotec). Both antibodies were incubated for 10 min at 4 °C and cells were washed twice with PBS (1% BSA) after each labeling step. Cells were analyzed using a MACSQuant® X flow cytometer (Miltenyi Biotec).
Evaluation of plasmid retention
Precultures of P. pastoris CBS7435PSD32 (see Table S3) were grown overnight under selective conditions and afterwards protein expression was induced as described above. ScFv-expressing cells were enriched through magnetic-activated cell sorting (MACS) to deplete non-displaying cells for subsequent subculturing. Therefore, up to 108 cells were labeled with 100 µl biotinylated anti-HA antibody followed by an incubation with anti-biotin microbeads (130–090-485, Miltenyi Biotec) for 15 min on ice. After two washing steps in PBS pH 7.2, 1% BSA, 2 mM EDTA, cells were loaded on a magnetic LS column and magnetically separated with the MidiMACS™ separator (Miltenyi Biotec). The positively enriched cells were used to inoculate selective and non-selective YPD media (with 100 µg/ml nourseothricin or without, respectively) and repeatedly passaged as recently described (Nakamura et al. 2018). Samples were taken every 24 h until 96 h of cultivation. For determining percentage of plasmid containing cells, approximately 100–1000 cells according to OD600 were plated onto selective and non-selective YPD plates. After 2 days of incubation, the percentage of plasmid containing cells was calculated. For determinining the surface expression, cell aliquots were induced and labeled with 10 µl anti-HA-APC and anti-c-myc-FITC antibody and analyzed by flow cytometry. Fluorescence signals of cells were analyzed by a MACSQuant® X flow cytometer (Miltenyi Biotec). To validate the plasmid character of pPIC6α-PSD79 (see Table S3), a PCR assay with one set of directly adjacent, diverging primer pairs, binding the pUC ori region of pPIC6α-PSD79, was designed (Table S1). This way, a PCR product of the same size as the predicted pPIC6α-PSD79 would validate its circular structure.
Results
High-throughput construction of episomal P. pastoris display vectors using a modular cloning system
In order to tether the expressed antibody fragments to the cell wall of P. pastoris, a PSD system was generated that uses anchor proteins with either glycosylphosphatidylinositol (GPI), such as SAG1 and SED1, or internal repeats for expression on the cell surface (Fig. 1a). In both display systems, an N-terminal antibody fragment was connected to a CWP by a (Gly4Ser)3 linker to improve its extracellular accessibility. For the immunological detection of surface-displayed antibody fragments, a HA and c-myc tag were sequentially attached at the C-terminus of the fragment (Fig. 1a, b). Fifteen P. pastoris- or S. cerevisiae-specific elements (five promoters, three signal peptides, three CWPs, one terminator, and three origins of replication) involved in the surface expression of proteins as well as plasmid maintenance in P. pastoris and three different antibody fragments to be expressed on the surface were selected for the initial design of the novel PSD system (Fig. 1c). A DoE custom design was used for the systematic screening of five multi-level categorical factors (Table 1). The number of constructs needed to identify main factors conferring highest surface expression could therefore be reduced from 406 possible combinations to below 90 runs, with each run requiring a unique factor combination cloned into the display vector. A total of 80 individual PSD constructs were subsequently used for the identification of genetic elements conferring highest surface expression (pPIC6α-PSD1-80, see Table S3). The cloning system we have developed relies on Golden Gate assembly and consists of five basic module types (promoters, signal peptides, antibody fragments, CWPs, and ARSs) with up to five different elements for each module (Fig. 2a). Each basic module type is flanked by individual fusion sites, which are identical for the different elements allowing the insertion of a fully assembled transcription unit and the ARS sequence in one step into the destination vector (Fig. 2a). Transformation of the P. pastoris CBS7435 strain with this set of vectors resulted in a cohort of strains harboring these individual constructs leading to varying surface expression levels of the antibody fragments used for analysis.

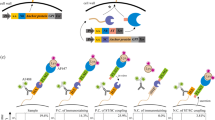
Modular cloning system for generation of PSD vectors and characterization of surface expression in P. pastoris. a Sets of basic modules contain synthesized genetic elements such as promoters (P), signal peptides (SP), antibody fragments (AF), cell wall proteins (CWP), and autonomously replicating sequences (ARS). A ribosome binding site (RBS), peptide tags, a linker (L), and an AOX1 transcription terminator (TT) were added to basic modules for functional replication, transcription and translation. PSD constructs are assembled from chosen basic modules in a one-pot and one-step cloning reaction. Basic modules of different types are flanked by compatible fusion sites. Each fusion site consists of 4 nucleotides of choice (boxed) flanked by a type IIS enzyme (BsaI) recognition site upstream and downstream of the DNA sequence (vertical box drawn under the fusion site). An exemplary assembled transcription unit for surface expression of fusion proteins is shown below. The ARS is located downstream of the transcription unit. b Exemplary PSD bearing P. pastoris cells (PSD ID11) were stained with fluorophore conjugated antibodies specific for c-myc (FITC) and HA (APC) tag and analyzed using flow cytometry. c Prediction profiler of the combined DoE analysis for all tested 80 PSD constructs. Constructs were simultaneously analyzed for the frequency of surface expression and stain indices for the two peptide tags. The red dashed vertical lines highlight selected set points for each of the experimental factors; selection affects the horizontal red dashed line which indicates surface expression based on the model prediction formula. Genetic elements with the highest desirability for all three experiment outputs are highlighted (red)
Identification of genetic elements conferring maximized surface expression
To test the effect of different genetic elements on antibody surface expression, displayed proteins were detected and analyzed by flow cytometry. The intensity of the two fluorescence signals and the frequency of cells expressing the fusion protein were evaluated based on the immunofluorescence labelling of the two peptide tags (Fig. 1a and b) as demonstrated for P. pastoris cells harboring PSD construct ID11 (Fig. 2b, see Table S3). This exemplary result of P. pastoris cells transformed with the pPIC6α-PSD11 vector showed a frequency of 17% fusion protein–displaying cells (Fig. 2b). The cell frequency of displayed fusion proteins, which can be used as a measure for the distribution of surface expression across a whole population, was determined for each construct as well as SIs for APC and FITC fluorescence, which assess the expression strength of fusion proteins (see Table S3). For the model proteins used in this study, a maximum display efficiency (percentage of displaying cells) of 25% for the anti-6 × histidine VHH, 17% for the anti-human CD123 scFv, and 15% for the anti-human IgG Fab region scFv were achieved, respectively. An MLR model was fitted for each flow cytometry output and model parameters are displayed in Table 2. All three models displayed a coefficient of determination (R2) of at least 0.5 and were highly significant (p-value < 0.0001), which is considered adequate for drawing conclusions from the models. Especially for the “frequency of cell expression” R2 was > 0.7, allowing more reliable predictions between the response (frequency of cell expression) and the explanatory variables. The models equally suggested that tested signal peptides were non-significant factors in the investigated combinations. Promoter, cell wall protein, ARS element, and antibody fragment were determined to be significant factors. As for the size effect (LogWorth, defined as -log10(p-value)) of each term in the models, the main factors promoter, ARS, and cell wall protein showed by far the strongest effects with values of 8.6 ± 1.9, 6.6 ± 4.1, and 6.0 ± 2.2, respectively (Fig. 2c).
The largest impact on display efficiency can be attributed to the promoters, with FLD1 and AOX1 resulting in significantly higher surface expression levels compared to the glycerol-inducible ADH1 promoter and constitutive promoters (AOD, ENO1). Overall, promoters FLD1 and AOX1 conferred approximately at least twofold higher expression compared to ADH1, AOD, and ENO1. The choice of cell wall protein and ARS sequence was found to have a significant impact on the surface expression as well, but to a lesser extent than promoter selection (Fig. 2c). The average difference in display efficiency between the least favorable PIR1 and the SAG1 anchor protein according to the prediction profiler was 1.6-fold and 1.4-fold between the mtDNA and panARS, respectively. In case of the signal peptides, all three elements performed similarly for the generated fusion proteins. The two scFvs with different specificities did show similar expression strength, but 1.3-fold lower display efficiency was observed when compared to the VHH fragment. Maximum surface expression was predicted for the combination of FLD1 promoter, SUC2 signal peptide, SAG1, VHH, and the panARS according to the prediction profiler and experimentally validated (Fig. 2c and Table S3).
Localization and estimation of surface displayed scFv molecules
Inhomogeneous surface expression of the antibody fragments may affect their interaction with the antigen. To confirm the uniform display of scFvs across the yeast surface, cells were further analyzed by confocal microscopy. Expression vectors encoding an anti-human CD123 scFv pYSDM1-CD123 and pPIC6α-PSD32 (Table 3) were introduced into S. cerevisiae EBY100 and P. pastoris CBS7435, respectively. In both anti-human CD123 scFv-expressing strains (S. cerevisiae EBY100CD123 and P. pastoris CBS7435CD123), fluorescence was homogeneously distributed on the cell surface, whereas no fluorescence was observed for control cells (Fig. 3). Sixty-nine percent of EBY100CD123 were displaying the anti-human CD123 scFv-α-agglutinin fusion protein which is 3.2-fold higher compared to CBS7435CD12. The MFI for scFv-displaying cells was reduced fourfold in PSD compared to YSD (23.4 ± 0.4 vs. 84.4 ± 5.0) indicating lower levels of display efficiency for PSD. The displayed molecule density of the P. pastoris CBS7435CD123 strain was further estimated by using a fluorescence quantitation. The measured MFI of approximately 25 (arbitrary units) detected for scFv-expressing CBS7435CD123 cells corresponds to an average number of 5 × 103 displayed molecules per cell (Fig. S1).

Fluorescence microscopy and flow cytometry analysis of P. pastoris and S. cerevisiae cells displaying single-chain variable fragment antibodies. Cells were labeled with an APC-conjugated antibody specific for the HA tag. Bright field (left column) and confocal microscopy using BP 620/60 filter after excitation at 633 nm (middle column) are shown. Marker bar = 10 µm. Flow cytometric analysis (right column) of anti-human CD123 scFv-displaying P. pastoris and S. cerevisiae cells. P. pastoris CBS7435 transformed with empty pPIC6α was used as control
Functional display of scFv fragments in P. pastoris
In order to evaluate the applicability of the generated PSD system and to demonstrate display of functional proteins on the cell surface, antigen binding of displayed anti-human CD123 scFvs in P. pastoris CBS7435CD123 was compared to S. cerevisiae EBY100CD123. When gated on scFv-expressing cells, binding capacities of P. pastoris and S. cerevisiae were similar with 80% and 84% of antigen binding cells, respectively (Fig. 4a). However, S. cerevisiae showed approximately a 1.5-fold higher functional display as determined by antigen binding than P. pastoris when compared to the vector controls. Additional PSD constructs ID38, ID40, and ID49 harboring different CWPs (SAG1 and SED1) were tested for CD123 antigen binding (Table 3). The frequency of antigen binding cells and fluorescence intensities varied across the constructs, despite similar surface expression levels (Fig. 4a). Cells harboring constructs with the SAG1 anchor (ID32 and ID49) showed a 1.3-fold higher frequency of antigen binding cells, ranging from 70 to 80%, compared to cells expressing SED1 anchor fusion proteins (present in ID38 and ID40, 53–60%). Additionally, a 2.4-fold higher functional display was observed for SAG1 anchor fusion proteins (present in ID 32 and ID49) compared to constructs carrying the SED1 anchor according to detected MFIs (present in ID38 and ID40, MFI of 2.6 ± 0.6 vs. 1.1 ± 0.2). Moreover, display of scFvs was tested for surface binding of spiked in Fabs. The displayed scFv was targeted against the Fab region of human antibodies and tested for binding towards 1 µM of two different Fab specificities, CD19 and CD33, using construct ID79 (Table 3, Fig. 4b). Comparable frequency of Fab binding cells was observed, with 65% (CD19 Fab) and 51% (CD33 Fab) binding and almost identical fold change of fluorescence signals in comparison to the control (~ 7 and ~ eightfold). Thus, the developed PSD system has shown, despite reduced display efficiency, similar functional surface expression of scFvs in comparison to YSD. Additionally, this display system is capable of capturing proteins of different protein classes demonstrating its potential broader applicability.

Functional binding studies of surface-expressed single-chain antibody fragments. Binding capabilities for two tested antigens (CD123 and Fab) displayed on the surface of P. pastoris and S. cerevisiae were determined by flow cytometry. Cells transformed with the vector control (negative control) are shown as unfilled histograms. Frequency of antigen binding cells and MFI-fold change compared to vector control for individual constructs are shown. a P. pastoris and S. cerevisiae expressing anti-human CD123 scFvs (upper lane) bound to biotinylated human CD123 and secondary labeled with an anti-biotin antibody conjugated with VioBlue. P. pastoris cells expressing anti-human CD123 scFvs in different genetic compositions (middle lane, Table 3) construct ID38 (left), ID40 (mid), or ID49 (right) were bound to biotinylated human CD123 and labeled with an anti-biotin antibody conjugated with VioBlue. b P. pastoris cells expressing construct ID79 (lower lane) bound to recombinant human Fab antibody fragments specific for CD19 (left) and CD33 (right) and labeled with a biotinylated anti-6 × histidine antibody and anti-biotin VioBlue. Typically 15–25% of the yeast population was displaying scFvs
Switchable display of antibody fragments
The use of episomal plasmids in our PSD system allows for the antibiotic-mediated control of plasmid maintenance and associated surface display of the antibody fragment fusion proteins. Thus, by removing the selective pressure, the surface expression of the antibody fragment fusion protein can be switched off on demand. A switchable surface display of antibody fragments could pave the way for a novel Fab/IgG secrete-and-capture approach which utilizes a transient capture of secreted proteins for fluorescence-activated cell sorting (FACS) of producer cells. The advantage of this approach lies in the fact that isolated cells may be used for immediate production purposes because the capturing moiety can be switched off in contrast to displayed antibody fragments expressed from integrative vectors.
Therefore, we have assessed the surface display level of an anti-human IgG Fab region scFv and the plasmid retention of its encoding vector in P. pastoris grown under selective and non-selective conditions. The P. pastoris CBS7435 strain was transformed with the pPIC6α-PSD79 vector, harboring the panARS sequence for plasmid retention. The PCR result in Fig. 5a visibly demonstrates the presence of a full length episomal circular plasmid of 5661 bp as shown in Fig. 1. The plasmid stability of pPIC6α-PSD79 was investigated by plating cells and estimating surface expression for the antibody fusion protein during repeated passaging over the course of 4 days (Fig. 5b). ScFv-displaying cells were enriched via MACS to deplete non-displaying cells for subsequent subculturing. The initial frequency of plasmid retention and scFv display of the cell population after MACS was 87% and 84%, respectively. After 96 h of cultivation, pPIC6α-PSD79 is stably maintained in up to approximately 60% of cells grown under selective conditions, while under non-selective conditions, the plasmid retention continually decreases to below 5% (Fig. 5b). When transformed cells were grown under nonselective conditions (i. e., without nourseothricin), low scFv display (around 4%) was observed at 96 h of cultivation accordingly (Fig. 5c). In contrast, approximately 45% of the cells grown under selective pressure showed scFv display with a wide range of fluorescence signals (Fig. 5c). Taken together, the plasmid stability and PCR assay clearly validated the plasmid character of pPIC6α-PSD79. The results indicate that panARS does not fully provide autonomous plasmid retention, such that almost half (40%) of the cells lost the plasmid during cultivation in presence of selective pressure at day 4 (Fig. 5b).

Evaluation of autonomously replicating activity and plasmid retention in P. pastoris. a PCR assay used to prove episomal presence of circular pPIC6α-PSD79 (5661 bp) in CBS7435. Lanes: (M) marker, relevant band sizes have been highlighted. (1) Amplicon generated with primer pair pUC ori-FW/RV. b Plasmid stability and scFv surface expression in P. pastoris, for repeated subcultures grown under selective (red) and non-selective (green) conditions. Plasmid retention was determined by plating cells on selective and non-selective agar plates. Error bars represent the standard deviation, with n = 2. (c) Flow cytometric analysis for scFv expression of cells grown under either selective (left) or non-selective (right) conditions after 96 h of cultivation. Cells were labeled with an APC-conjugated antibody specific for the HA tag. Frequency of antibody expressing cells compared to vector control is shown
Discussion
The developed antibody display in P. pastoris is based on circular plasmids containing ARS elements which allow episomal plasmid maintenance, but are inherently unstable in the absence of selective pressure. For the construction of episomal antibody display vectors, a variety of different genetic elements were evaluated for the applicability in PSD. Here, we have shown the rapid construction of a diverse set of vectors consisting of many different transcription units, including ARS elements, using Golden Gate cloning strategy (80 constructs, consisting of 17 individual basic modules). This set of constructs was systematically screened by employing a custom design to identify genetic elements conferring maximized surface expression. The goodness of fit for the separate models suggested the models to be suitable for the purpose of factor screening. Broken down into the individual module types, measured antibody display efficiencies for the methanol-inducible AOX1 and FLD1 promoters were higher compared to the glycerol-inducible ADH1 promoter and constitutive promoters (AOD, ENO1). This is well in line with their attributed promoter strengths described in literature (Vogl and Glieder 2013). Among the tested CWPs, the GPI proteins (SAG1, SED1) were most effective in displaying antibody fragments compared to PIR1. Even though PIR1 was used for successful display of enzymes and reporter proteins in P. pastoris (Moura et al. 2015; Yang et al. 2017), studies have reported considerable variations in the formation of specific protein-polysaccharide complexes depending on several environmental factors in S. cerevisiae, which could be an explanation for the results of our screening (Klis et al. 2002). When comparing display efficiency of SED1 anchor proteins to the a-agglutinin anchor system, which like PIR1 also relies on the formation of disulfide bonds, a higher efficiency for the GPI anchor in S. cerevisiae was reported by Yang et al. (2019).
In regard to the ARS elements tested in this study, highest display levels were achieved with panARS-based expression vectors. ARSs generally serve as the origin of DNA replication and are closely related to stability, transformation efficiency, and copy number of episomal plasmids (Pena et al. 2018). Therefore, increased surface display can probably be attributed to higher copy numbers and higher plasmid stability of episomal vectors containing the panARS element (Camattari et al. 2016; Liachko and Dunham 2014), since vectors harboring either PARS1 or mtDNA have resulted in lower copy numbers (Nakamura et al. 2018; Schwarzhans et al. 2017). In fact, higher expression efficiency of a model protein has been shown to be correlated to the copy number of plasmids containing the panARS (Camattari et al. 2016). A systematic comparison of abovementioned ARSs used in a CRISPR/Cas9 genome editing system in P. pastoris confirmed highest efficiency for panARS-based vectors owing to their higher plasmid stability and copy number (Gu et al. 2019). The proportion of cells displaying antibody fragments from an integrated expression vector with 67% is approximately threefold higher compared to our results of around 21% cell frequency (Ryckaert et al. 2010). As expected, the significant difference in expressors and non-expressors further indicates that the plasmid stability is an important factor for the efficiency of protein expression. Vectors that are integrated into the genome of P. pastoris typically result in stable expression strains even under non-selective conditions (Cregg et al. 2000), thus resulting in a higher frequency of an expressing population. Other effects related to vector integration, such as the integration site and genome rearrangements, may influence protein expression as well (Aw et al. 2017; Love et al. 2010), but do not apply to strains harboring episomal plasmids.
To our knowledge, antibody fragment display facilitated by episomal vectors in P. pastoris has not been reported so far, but intracellular expression of fluorescent reporter proteins from episomal plasmids achieved frequencies which range from 40 to 65% and were 2–threefold higher than the display efficiency determined in this study (Nakamura et al. 2018; Gu et al. 2019). Unlike intracellularly expressed proteins, secreted proteins have to undergo membrane translocation, signal peptide processing, and folding within the ER, which have been identified as major bottlenecks for recombination protein production in P. pastoris (reviewed by Puxbaum et al. 2015).
Surface display in S. cerevisiae and P. pastoris were both based on episomal plasmids which make use of strong promoters. The frequency of antibody displaying cells is higher in YSD compared to PSD, but two clearly distinct populations of expressing and non-expressing cells were present in both display systems. The observed display efficiency of 69% in YSD was similar to those efficiencies described in the literature (Kuroda et al. 2009). In addition, antibody display can be further evaluated by estimation of displayed molecule density. It has been shown that the density of expressed proteins on the cell surface by using integrative vectors and the α-agglutinin anchor in P. pastoris ranges from 4 to 9 × 105 molecules (Jin et al. 2014; Mergler et al. 2004). However, when the POI is expressed from an episomal vector in YSD, the number of proteins per cell for the α-agglutinin anchor was estimated at 2.5 × 104 and 1.5 × 104 (Kato et al. 2007; Shibasaki et al. 2001), indicating that the number of molecules displayed on the cell surface is generally reduced when using episomal instead of integrative vectors. The amount of displayed POI in the developed PSD system with 5 × 103 molecules per cell is approximately 3- to fivefold lower than determined in the studies of Shibasaki et al. (2001) and Kato et al. (2007). Quantification of the MFI of S. cerevisiae and P. pastoris cells expressing scFvs, an indirect measure for displayed molecules per cell, confirmed lower display levels in PSD compared to YSD. While previous studies have shown that for PARS1 and mtDNA approximately 50–55% of the cells lost the plasmid during culturing under selective conditions (Nakamura et al. 2018; Schwarzhans et al. 2017), similar stability rates for plasmids harboring the panARS were observed. The plasmid loss rate in S. cerevisiae is significantly reduced in comparison to P. pastoris. Expression of a GFP-carrying plasmid resulted in the emission of strong fluorescence from 80% of the cell population when cultivated under selective conditions (Hegemann et al. 1999). Episomal plasmids in S. cerevisiae bear an additional centromeric (CEN) sequence besides the ARS. CEN sequences act as attachment sites for kinetochore complexes, which are involved in the chromosome segregation and enable a high segregational stability during mitosis (Westermann et al. 2007). As demonstrated for haploid S. cerevisiae strains, CEN6/ARS4 plasmids are maintained at a low copy number, ranging from 2 to 5 copies per cell (Karim et al. 2013), which is lower than the determined copy number of 6 to 17 copies per cell for panARS vectors (Camattari et al. 2016; Gu et al., 2019). Thus, suggesting that the plasmid instability of episomal vectors in P. pastoris stems from poor plasmid segregation rather than low replication activity. The plasmid stability achieved in a recent study supports this assumption, since up to 84% of cells were able to maintain episomal plasmids of one copy per cell when a full-length centromere was used as an ARS (Nakamura et al. 2018). Therefore, the differences in expression efficiency of surface-displayed scFvs between YSD and PSD as indicated by lower cell frequency and MFI are most likely caused by insufficient plasmid retention (autonomous replication activity, plasmid replication, and/or partitioning) for the used ARSs in P. pastoris when to compared S. cerevisiae.
In addition to the employed promoters, copy number, and stability of used plasmids, varying surface expression of fusion proteins between PSD and YSD could also be ascribed to their structural and biochemical/metabolic differences. Although S. cerevisiae and P. pastoris both belong to the Saccharomycetaceae family, the Golgi apparatus of S. cerevisiae for example differs from its counterpart in P. pastoris to some extent, which is rather resembling the structural organization of the Golgi apparatus of higher eukaryotic and human cells (Mogelsvang et al. 2003; Rossanese et al. 1999). This difference might especially affect the glycosylation, secretion and cell wall composition of the two yeasts. Moreover, most genetic elements employed for surface display in P. pastoris in this study were obtained from endogenous sequences of S. cerevisiae and have not been codon-optimized for expression in P. pastoris. A less favorable codon bias might have negatively influenced the transcription and translation efficiency and led to lower expression levels (Tuller et al. 2010; Xu et al. 2021).
Nonetheless, results from fluorescence microscopy and flow cytometry confirmed the successful display of antibody fragments on the cell surface of P. pastoris. Regarding the functional display of antibody fragments, we have demonstrated comparable antigen binding capability of an anti-human CD123 scFv antibody for the developed PSD, which utilizes the α-agglutinin anchor protein compared to the antibody display in S. cerevisiae. Fluorescence microscopy confirmed homogenous expression of fusion proteins across the cell surface for both display systems. Interestingly, alternating genetic compositions, especially in case of anchor proteins, resulted in varying antigen binding cell frequencies and varying amounts of surface bound antigen. Ikonuma et al. (2020) have demonstrated that the anchorage position of GPI-attached proteins in the cell wall could be controlled by changing the fused anchoring domain for SED1 and SAG1 in S. cerevisiae, indicating that the observed difference in antigen binding can be attributed to the surface positioning of the POI, which in this study was more favorable for SAG1-fused scFvs.
Future strategies towards improving antibody display may focus on a multitude of different aspects. For example, signal peptide optimization and engineering of a hybrid secretion signal have previously enhanced the secretion of proteins in P. pastoris (Aw et al. 2018; Barrero et al. 2018). By employing an endogenous centromere sequence in P. pastoris, plasmid retention rates similar to S. cerevisiae were achieved (Nakamura et al. 2018). However, due to the size of the sequence (6655 bp), resulting plasmids will be very large affecting e.g. cloning and/or transformation efficiencies. The use of ARS sequences in P. pastoris derived from other yeasts has also allowed for stable plasmid maintenance in transformed cells (Vogl 2015). New ARSs and plasmid partitioning elements have been identified in a high-throughput manner in S. cerevisiae (Hoggard et al. 2016; Liachko et al. 2013). This approach could be applied to other non-conventional yeasts in order to identify additional ARSs for potential use in P. pastoris. Mutating existing ARSs was shown improve their function and resulted in higher plasmid stability in yeast as well (Liachko et al. 2013, 2014). Even the growth rate can influence the stability of yeast plasmids (Mead et al. 1986). Therefore, employing a P. pastoris strain that does not contain any functional AOX gene might be beneficial in regard to plasmid loss. For this methanol utilization negative strain very little growth was observed in the presence of methanol when compared to the mutS phenotype (Zavec et al. 2020). Reduced proliferation might thereby potentially translate to higher plasmid retention.
Lastly, plasmid retention and protein retention were monitored independently by measuring the retention of the plasmid-borne selectable marker and labelling of surface expressed proteins during growth in non-selective media. Both plasmid loss and protein loss for cells grown under non-selective conditions were confirmed, implying the cells’ ability to be cured of the plasmids during cultivation in the absence of selective pressure. In this study, we established and characterized a novel system for the functional display of antibody fragments on the surface of P. pastoris. Besides surface binding of a clinically relevant antigen, two Fab antibody fragments were successfully captured on the surface of P. pastoris, demonstrating that the PSD system is capable of displaying different protein classes. In combination with the transient display of antibody fragments, the latter has the potential for a secrete-and-capture method for the isolation of antibody producing yeast clones via FACS as outlined before (Rakestraw et al. 2011; Rhiel et al. 2014), but with the advantage that selected clones are suitable for immediate use in production processes (Gätjen et al., manuscript in preparation). In general, antibody engineering in this commonly used expression host would offer the possibility to directly select antibodies in the same organism used for subsequent production, facilitating and optimizing clone selection processes. However, PSD based on episomal plasmids still needs further improvement to be able to compete with the efficiency of YSD.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Andreu C, del Olmo ML (2018) Yeast arming systems: pros and cons of different protein anchors and other elements required for display. Appl Microbiol Biotechnol 102:2543–2561. https://doi.org/10.1007/s00253-018-8827-6
Aw R, Barton GR, Leak DJ (2017) Insights into the prevalence and underlying causes of clonal variation through transcriptomic analysis in Pichia pastoris. Appl Microbiol Biotechnol 101:5045–5058. https://doi.org/10.1007/s00253-017-8317-2
Aw R, McKay PF, Shattock RJ, Polizzi KM (2018) A systematic analysis of the expression of the anti-HIV VRC01 antibody in Pichia pastoris through signal peptide optimization. Protein Expr Purif 149:43–50. https://doi.org/10.1016/j.pep.2018.03.013
Barrero JJ, Casler JC, Valero F, Ferrer P, Glick BS (2018) An improved secretion signal enhances the secretion of model proteins from Pichia pastoris. Microb Cell Fact 17:1–13. https://doi.org/10.1186/s12934-018-1009-5
Boder ET, Wittrup KD (1997) Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol 15:553–557. https://doi.org/10.1038/nbt0697-553
Brown SR, Staff M, Lee R, Love J, Parker DA, Aves SJ, Howard TP (2018) Design of experiments methodology to build a multifactorial statistical model describing the metabolic interactions of alcohol dehydrogenase isozymes in the ethanol biosynthetic pathway of the yeast Saccharomyces cerevisiae. ACS Synth Biol 7:1676–1684. https://doi.org/10.1021/acssynbio.8b00112
Camattari A, Goh A, Yip LY, Tan AHM, Ng SW, Tran A, Liu G, Liachko I, Dunham MJ, Rancati G (2016) Characterization of a panARS-based episomal vector in the methylotrophic yeast Pichia pastoris for recombinant protein production and synthetic biology applications. Microb Cell Fact 15:1–11. https://doi.org/10.1186/s12934-016-0540-5
Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD (2006) Isolating and engineering human antibodies using yeast surface display. Nat Protoc 1:755–768. https://doi.org/10.1038/nprot.2006.94
Cherf GM, Cochran JR (2015) Applications of yeast surface display for protein engineering. Methods Mol Biol 1319:155–175. https://doi.org/10.1007/978-1-4939-2748-7_8
Cregg JM, Barringer KJ, Hessler AY, Madden K (1985) Pichia pastoris as a host system for transformations. Mol Cell Biol 5:3376–3385. https://doi.org/10.1128/mcb.5.12.3376-3385.1985
Cregg JM, Cereghino JL, Shi J, Higgins DR (2000) Recombinant protein expression in Pichia pastoris. Mol Biotechnol 16:23–52. https://doi.org/10.1385/MB:16:1:23
Engler C, Kandzia R, Marillonnet S (2008) A one pot, one step, precision cloning method with high throughput capability. PLoS ONE 3:e3647. https://doi.org/10.1371/journal.pone.0003647
Gai SA, Wittrup KD (2007) Yeast surface display for protein engineering and characterization. Curr Opin Struct Biol 17:467–473. https://doi.org/10.1016/j.sbi.2007.08.012
Gu Y, Gao J, Cao M, Dong C, Lian J, Huang L, Cai J, Xu Z (2019) Construction of a series of episomal plasmids and their application in the development of an efficient CRISPR/Cas9 system in Pichia pastoris. World J Microbiol Biotechnol 35:1–10. https://doi.org/10.1007/s11274-019-2654-5
Hegemann JH, Klein S, Heck S, Güldener U, Niedenthal RK, Fleig U (1999) A fast method to diagnose chromosome and plasmid loss in Saccharomyces cerevisiae strains. Yeast 15:1009–1019. https://doi.org/10.1002/(SICI)1097-0061(199907)15:10B%3c1009::AID-YEA396%3e3.0.CO;2-I
Hoggard T, Liachko I, Burt C, Meikle T, Jiang K, Craciun G, Dunham MJ, Fox CA (2016) High throughput analyses of budding yeast ARSs reveal new DNA elements capable of conferring centromere-independent plasmid propagation. G3 (Bethesda) 6:993–1012. https://doi.org/10.1534/g3.116.027904
Inokuma K, Kurono H, den Haan R, van Zyl WH, Hasunuma T, Kondo A (2020) Novel strategy for anchorage position control of GPI-attached proteins in the yeast cell wall using different GPI-anchoring domains. Metab Eng 57:110–117. https://doi.org/10.1016/j.ymben.2019.11.004
Jin Z, Zhang K, Zhang L, Zheng SP, Han SY, Lin Y (2014) Quantification analysis of yeast-displayed lipase. Anal Biochem 450:46–48. https://doi.org/10.1016/j.ab.2013.12.035
Karbalaei M, Rezaee SA, Farsiani H (2020) Pichia pastoris: a highly successful expression system for optimal synthesis of heterologous proteins. J Cell Physiol 235:5867–5881. https://doi.org/10.1002/jcp.29583
Karim AS, Curran KA, Alper HS (2013) Characterization of plasmid burden and copy number in Saccharomyces cerevisiae for optimization of metabolic engineering applications. FEMS Yeast Res 13:107–116. https://doi.org/10.1111/1567-1364.12016
Kato M, Fuchimoto J, Tanino T, Kondo A, Fukuda H, Ueda M (2007) Preparation of a whole-cell biocatalyst of mutated Candida antarctica lipase B (mCALB) by a yeast molecular display system and its practical properties. Appl Microbiol Biotechnol 75:549–555. https://doi.org/10.1007/s00253-006-0835-2
Khasa Y, Conrad S, Sengul M, Plautz S, Meagher MM, Inan M (2011) Isolation of Pichia pastoris PIR genes and their utilization for cell surface display and recombinant protein secretion. Yeast 28:213–226. https://doi.org/10.1002/yea.1832
Kim H, Yoo SJ, Kang HA (2015) Yeast synthetic biology for the production of recombinant therapeutic proteins. FEMS Yeast Res 15:1–16. https://doi.org/10.1111/1567-1364.12195
Klis FM, Mol P, Hellingwerf K, Brul S (2002) Dynamics of cell wall structure in Saccharomyces cerevisiae. FEMS Microbiol Rev 26:239–256. https://doi.org/10.1111/j.1574-6976.2002.tb00613.x
Kuroda K, Matsui K, Higuchi S, Kotaka A, Sahara H, Hata Y, Ueda M (2009) Enhancement of display efficiency in yeast display system by vector engineering and gene disruption. Appl Microbiol Biotechnol 82:713-719. https://doi.org/10.1007/s00253-008-1808-4
Lee CC, Williams TG, Wong DW, Robertson GH (2005) An episomal expression vector for screening mutant gene libraries in Pichia pastoris. Plasmid 54:80–85. https://doi.org/10.1016/j.plasmid.2004.12.001
Li H, Sethuraman N, Stadheim TA, Zha D, Prinz B, Ballew N, Bobrowicz P, Choi BK, Cook WJ, Cukan M, Houston-Cummings NR, Davidson R, Gong B, Hamilton SR, Hoopes JP, Jiang Y, Kim N, Mansfield R, Nett JH, Rios S, Strawbridge R, Wildt S, Gerngross TU (2006) Optimization of humanized IgGs in glycoengineered Pichia pastoris. Nat Biotechnol 24:210–215. https://doi.org/10.1038/nbt1178
Liachko I, Youngblood RA, Keich U, Dunham MJ (2013) High-resolution mapping, characterization, and optimization of autonomously replicating sequences in yeast. Genome Res 23:698–704. https://doi.org/10.1101/gr.144659.112
Liachko I, Dunham MJ (2014) An autonomously replicating sequence for use in a wide range of budding yeasts. FEMS Yeast Res 14:364–367. https://doi.org/10.1111/1567-1364.12123
Liachko I, Youngblood RA, Tsui K, Bubb KL, Queitsch C, Raghuraman MK, Nislow C, Brewer BJ, Dunham MJ (2014) GC-rich DNA elements enable replication origin activity in the methylotrophic yeast Pichia pastoris. PLoS Genet 10:e1004169. https://doi.org/10.1371/journal.pgen.1004169
Lin S, Houston-Cummings NR, Prinz B, Moore R, Bobrowicz B, Davidson RC, Wildt S, Stadheim TA, Zha D (2012) A novel fragment of antigen binding (Fab) surface display platform using glycoengineered Pichia pastoris. Journal I Methods 375:159–165. https://doi.org/10.1016/j.jim.2011.10.003
Liu L, Gomathinayagam S, Hamuro L, Prueksaritanont T, Wang W, Stadheim TA, Hamilton SR (2013) The impact of glycosylation on the pharmacokinetics of a TNFR2: Fc fusion protein expressed in glycoengineered Pichia pastoris. Pharm Res 30:803–812. https://doi.org/10.1007/s11095-012-0921-3
Love KR, Panagiotou V, Jiang B, Stadheim TA, Love JC (2010) Integrated single-cell analysis shows Pichia pastoris secretes protein stochastically. Biotechnol Bioeng 106:319–325. https://doi.org/10.1002/bit.22688
McCafferty J, Griffiths AD, Winter G, Chiswell DJ (1990) Phage antibodies: filamentous phage displaying antibody variable domains. Nature 348:552–554. https://doi.org/10.1038/348552a0
Mead DJ, Gardner DC, Oliver SG (1986) The yeast 2 μ plasmid: strategies for the survival of a selfish DNA. Mol Gen Genet 205:417–421. https://doi.org/10.1007/BF00338076
Mergler M, Wolf K, Zimmermann M (2004) Development of a bisphenol A-adsorbing yeast by surface display of the Kluyveromyces yellow enzyme on Pichia pastoris. Appl Microbiol Biotechnol 63:418–421. https://doi.org/10.1007/s00253-003-1361-0
Mogelsvang S, Gomez-Ospina N, Soderholm J, Glick BS, Staehelin LA (2003) Tomographic evidence for continuous turnover of Golgi cisternae in Pichia pastoris. Mol Biol Cell 14:2277–2291. https://doi.org/10.1091/mbc.e02-10-0697
Montgomery DC (2013) Design and analysis of experiments. John Wiley & Sons Inc, Hoboken, NJ
Moura MVH, da Silva GP, Machado ACDO, Torres FAG, Freire DMG, Almeida RV (2015) Displaying lipase B from Candida antarctica in Pichia pastoris using the yeast surface display approach: prospection of a new anchor and characterization of the whole cell biocatalyst. PLoS ONE 10:e0141454. https://doi.org/10.1371/journal.pone.0141454
Näätsaari L, Mistlberger B, Ruth C, Hajek T, Hartner FS, Glieder A (2012) Deletion of the Pichia pastoris KU70 homologue facilitates platform strain generation for gene expression and synthetic biology. PLoS ONE 7:e39720. https://doi.org/10.1371/journal.pone.0039720
Nakamura Y, Nishi T, Noguchi R, Ito Y, Watanabe T, Nishiyama T, Aikawa S, Hasunuma T, Ishii J, Okubo Y, Kondo A (2018) A stable, autonomously replicating plasmid vector containing Pichia pastoris centromeric DNA. Appl Environ Microbiol 84:e02882-e2917. https://doi.org/10.1128/AEM.02882-17
Obst U, Lu TK, Sieber V (2017) A modular toolkit for generating Pichia pastoris secretion libraries. ACS Synth Biol 6:1016–1025. https://doi.org/10.1021/acssynbio.6b00337
Peña DA, Gasser B, Zanghellini J, Steiger MG, Mattanovich D (2018) Metabolic engineering of Pichia pastoris. Metab Eng 50:2–15. https://doi.org/10.1016/j.ymben.2018.04.017
Puxbaum V, Mattanovich D, Gasser B (2015) Quo vadis? The challenges of recombinant protein folding and secretion in Pichia pastoris. Appl Microbiol Biotechnol 99:2925–2938. https://doi.org/10.1007/s00253-015-6470-z
Rakestraw JA, Aird D, Aha PM, Baynes BM, Lipovšek D (2011) Secretion-and-capture cell-surface display for selection of target-binding proteins. Protein Eng Des Sel 24:525–530. https://doi.org/10.1093/protein/gzr008
Rhiel L, Krah S, Günther R, Becker S, Kolmar H, Hock B (2014) REAL-Select: full-length antibody display and library screening by surface capture on yeast cells. PLoS ONE 9:e114887. https://doi.org/10.1371/journal.pone.0114887
Rossanese OW, Soderholm J, Bevis BJ, Sears IB, O’Connor J, Williamson EK, Glick BS (1999) Golgi structure correlates with transitional endoplasmic reticulum organization in Pichia pastoris and Saccharomyces cerevisiae. J Cell Biol 145:69–81. https://doi.org/10.1083/jcb.145.1.69
Ryckaert S, Martens V, De Vusser K, Contreras R (2005) Development of a S. cerevisiae whole cell biocatalyst for in vitro sialylation of oligosaccharides. J Biotechnol 119:379–388. https://doi.org/10.1016/j.jbiotec.2005.04.010
Ryckaert S, Pardon E, Steyaert J, Callewaert N (2010) Isolation of antigen-binding camelid heavy chain antibody fragments (nanobodies) from an immune library displayed on the surface of Pichia pastoris. J Biotechnol 145:93–98. https://doi.org/10.1016/j.jbiotec.2009.10.010
Sasagawa T, Matsui M, Kobayashi Y, Otagiri M, Moriya S, Sakamoto Y, Ito Y, Lee CC, Kitamoto K, Arioka M (2011) High-throughput recombinant gene expression systems in Pichia pastoris using newly developed plasmid vectors. Plasmid 65:65–69. https://doi.org/10.1016/j.plasmid.2010.08.004
Schwarzhans JP, Luttermann T, Wibberg D, Winkler A, Hübner W, Huser T, Kalinowski J, Friehs K (2017) A mitochondrial autonomously replicating sequence from Pichia pastoris for uniform high level recombinant protein production. Front Microbiol 8:780. https://doi.org/10.3389/fmicb.2017.00780
Scott JK, Smith GP (1990) Searching for peptide ligands with an epitope library. Science 249:386–390. https://doi.org/10.1126/science.1696028
Shaheen HH, Prinz B, Chen MT, Pavoor T, Lin S, Houston-Cummings NR, Moore R, Stadheim TA, Zha D (2013) A dual-mode surface display system for the maturation and production of monoclonal antibodies in glyco-engineered Pichia pastoris. PLoS ONE 8:e70190. https://doi.org/10.1371/journal.pone.0070190
Shibasaki S, Ueda M, Iizuka T, Hirayama M, Ikeda Y, Kamasawa N, Osumi M, Tanaka A (2001) Quantitative evaluation of the enhanced green fluorescent protein displayed on the cell surface of Saccharomyces cerevisiae by fluorometric and confocal laser scanning microscopic analyses. Appl Microbiol Biotechnol 55:471–475. https://doi.org/10.1007/s002530000539
Ståhl S, Uhlén M (1997) Bacterial surface display: trends and progress. Trends Biotechnol 15:185–192. https://doi.org/10.1016/S0167-7799(97)01034-2
Su GD, Zhang X, Lin Y (2010) Surface display of active lipase in Pichia pastoris using Sed1 as an anchor protein. Biotechnol Lett 32:1131–1136. https://doi.org/10.1007/s10529-010-0270-4
Tanino T, Ohno T, Aoki T, Fukuda H, Kondo A (2007) Development of yeast cells displaying Candida antarctica lipase B and their application to ester synthesis reaction. Appl Microbiol Biotechnol 75:1319–1325. https://doi.org/10.1007/s00253-007-0959-z
Tang H, Wang S, Wang J, Song M, Xu M, Zhang M, Shen Y, Hou J, Bao, X (2016) N-hypermannose glycosylation disruption enhances recombinant protein production by regulating secretory pathway and cell wall integrity in Saccharomyces cerevisiae. Sci Rep 6:1-13. https://doi.org/10.1038/srep25654
Tuller T, Waldman YY, Kupiec M, Ruppin E (2010) Translation efficiency is determined by both codon bias and folding energy. Proc Natl Acad Sci U S A 107:3645–3650. https://doi.org/10.1073/pnas.0909910107
Vervecken W, Kaigorodov V, Callewaert N, Geysens S, De Vusser K, Contreras R (2004) In vivo synthesis of mammalian-like, hybrid-type N-glycans in Pichia pastoris. Appl Environ Microbiol 70:2639–2646. https://doi.org/10.1128/AEM.70.5.2639-2646.2004
Vogl T, Glieder A (2013) Regulation of Pichia pastoris promoters and its consequences for protein production. N Biotechnol 30:385–404. https://doi.org/10.1016/j.nbt.2012.11.010
Vogl T (2015) Synthetic biology to improve protein expression in Pichia pastoris. Dissertation, Graz University of Technology. https://diglib.tugraz.at/synthetic-biology-to-improve-protein-expression-in-pichia-pastoris-2015. Accessed 29 June 2022
Weis R, Luiten R, Skranc W, Schwab H, Wubbolts M, Glieder A (2004) Reliable high-throughput screening with Pichia pastoris by limiting yeast cell death phenomena. FEMS Yeast Res 5:179–189. https://doi.org/10.1016/j.femsyr.2004.06.016
Weninger A, Hatzl AM, Schmid C, Vogl T, Glieder A (2016) Combinatorial optimization of CRISPR/Cas9 expression enables precision genome engineering in the methylotrophic yeast Pichia pastoris. J Biotechnol 235:139–149. https://doi.org/10.1016/j.jbiotec.2016.03.027
Westermann S, Drubin DG, Barnes G (2007) Structures and functions of yeast kinetochore complexes. Annu Rev Biochem 76:563–591. https://doi.org/10.1146/annurev.biochem.76.052705.160607
Xu Y, Liu K, Han Y, Xing Y, Zhang Y, Yang Q, Zhou M (2021) Codon usage bias regulates gene expression and protein conformation in yeast expression system P. pastoris. Microb Cell Fact 20:1–15. https://doi.org/10.1186/s12934-021-01580-9
Yang S, Lv X, Wang X, Wang J, Wang R, Wang T (2017) Cell-surface displayed expression of trehalose synthase from Pseudomonas putida ATCC 47054 in Pichia pastoris using Pir1p as an anchor protein. Front Microbiol 8:2583. https://doi.org/10.3389/fmicb.2017.02583
Yang X, Tang H, Song M, Shen Y, Hou J, Bao X (2019) Development of novel surface display platforms for anchoring heterologous proteins in Saccharomyces cerevisiae. Microb Cell Fact 18:1–10. https://doi.org/10.1186/s12934-019-1133-xFigures
Zavec D, Gasser B, Mattanovich D (2020) Characterization of methanol utilization negative Pichia pastoris for secreted protein production: New cultivation strategies for current and future applications. Biotechnol Bioeng 117:1394–1405. https://doi.org/10.1002/bit.27303
Acknowledgements
We would like to thank Jan Drewes (Miltenyi Biotec) for establishing various laboratory automation protocols and providing useful input during troubleshooting. In addition, we are grateful to Marius Terfrüchte, Alexandra Schmitz, and Sven Karstaedt (Miltenyi Biotec) for the transfer of P. pastoris cells and for supporting the work in this study.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: DG, FT and JCD. Performed the experiments: DG. Analyzed the data: DG, FT, JCD, MD, and VN. Wrote the paper: DG and MD. Reviewed and edited the manuscript: VN and MW. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Competing interests
DG, FT, JCD, MD, VN, and MW are employees of Miltenyi Biotec B.V. & Co. KG. DG has relevant IP to the findings disclosed. No relevant financial or non-financial interests exist for the remaining authors.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gätjen, D., Tomszak, F., Dettmann, JC. et al. Design of a novel switchable antibody display system in Pichia pastoris. Appl Microbiol Biotechnol 106, 6209–6224 (2022). https://doi.org/10.1007/s00253-022-12108-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-022-12108-5