
Abstract
The gene encoding a Baeyer-Villiger monooxygenase and identified in Pseudomonas putida KT2440 was cloned and functionally expressed in Escherichia coli. The highest yield of soluble protein could be achieved by co-expression of molecular chaperones. In order to determine the substrate specificity, biocatalyses were performed using crude cell extract, growing and resting cells. Examination of aromatic, cyclic and aliphatic ketones revealed a high specificity towards short-chain aliphatic ketones. Interestingly, some open-chain ketones were converted to the alkylacetates, while for others formation of the ester products with oxygen on the other side of the keto group could also be detected yielding the corresponding methyl or ethyl esters.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Baeyer-Villiger monooxygenases (BVMOs) belong to the class of oxidoreductases and convert aliphatic, cyclic and aromatic ketones into esters and lactones, respectively, using molecular oxygen (Mihovilovic et al. 2002). They are flavin-dependent (mostly FAD) and require NAD(P)H to catalyze this reaction. The cyclohexanone monooxygenase from Acinetobacter calcoaceticus NCIMB 9871, converting mono- and bicyclic ketones (Donoghue et al. 1976, Stewart 1998), is the best studied BVMO so far. Other BVMOs converting aromatic ketones (Kamerbeek et al. 2001, Tanner and Hopper 2000) or aliphatic open-chain ketones (Britton and Markovetz 1977, Malito et al. 2004) were also described. Recently, the gene of a BVMO from Pseudomonas fluorescens DSM 50106 was cloned and functionally expressed in E. coli (Kirschner et al. 2007). This enzyme preferentially accepts aliphatic acyclic 2-ketones and converts racemic 4-hydroxy-2-ketones with good enantioselectivities (Kirschner and Bornscheuer 2006). In 2004 the first crystal structure of a BVMO from Thermobifida fusca, the phenylacetone monooxygenase, was published (Malito et al. 2004).
In 2002 the complete genome sequence of Pseudomonas putida KT2440 was published (GenBank accession number: AE015451) (Nelson et al. 2002). 15 open reading frames encoding putative monooxygenases were determined. Sequence analysis on protein and nucleotide level revealed one monooxygenase containing sequence motifs typical for Baeyer-Villiger monooxygenases (consensus sequence (FXGXXXHXXXW) and binding sites for cofactor and prosthetic group (GXGXXG-motifs)) (Fraaije et al. 2002). In this paper, we now describe the cloning, expression and the biocatalytical investigation of this newly found Baeyer-Villiger monooxygenase.
Materials and methods
Bacterial strains and plasmids
E. coli DH5α was from Clontech, E. coli JM109 was received from New England Biolabs, E. coli BL21 (DE3) and pET22b(+) were purchased from Novagen. Pseudomonas putida KT2440 was obtained from the FZ Jülich (Jülich, Germany). The Chaperone Plasmid Set containing the plasmids pG-KJE8, pGro7, pKJE7, pG-Tf2 and pTf16 was purchased from TaKaRa Bio Inc.
Construction of expression vectors
The BVMO gene was amplified from the genomic DNA from Ps. putida KT2440 without the stop codon using the polymerase chain reaction (PCR) with the oligonucleotides FW-PpKT2440-gDNA (5′-GTA CAT CGA TGG GAT CCT CGG CGG-3′) and RV-PpKT2440-gDNA (5′-GTA CGA GCT CCC ATA TGT CCT CTC ACA C-3′) and Pwo DNA polymerase (Roche). The restriction sites for NdeI and BamHI (underlined) in the primer sequences were used to perform a directed cloning between the NdeI/BamHI sites of the vector pJOE4072.6 (Kirschner et al. 2007). Hereby the BVMO gene was fused at the C-terminal end in frame to the six histidine codons (coding for a His-tag) following the BamHI site in the vector. This plasmid was named pJOE-KT2440. For cloning into pET22b(+), a HindIII restriction site within the gene was deleted using QuikChange site-directed mutagenesis (Stratagene). The mutated pJOE-KT2440 was then digested with NdeI and HindIII and the resulting fragment, consisting of the BVMO gene and the His-tag, was cloned into pET22b(+). This construct was named pET22b(+)-KT2440. Constructs without C-terminal His-tags were obtained by introducing a TGA stop-codon into the BamHI site using QuikChange site-directed mutagenesis.
Gene expression
E. coli JM109 was transformed with the pJOE construct, BL21 (DE3) with the pET22b(+) construct. In both cases expression was performed in 30 ml LB media containing 100 μg ampicillin/ml (LBamp) at 25, 30 and 37°C. At an optical density (600 nm) of 0.6–0.7, BVMO expression was induced by the addition of l-rhamnose (0.2% w/v final concentration) for the pJOE construct and with 0.5 mM IPTG for the pET22b(+) construct. During growth for up to 24 h, samples were taken, centrifuged, resuspended in 200 μl sodium phosphate buffer (50 mM, pH 7.5) and disrupted on ice by sonication (Sonoplus, Bandelin, Germany; 50% pulse and 50% amplitude). Inclusion bodies were separated from the cell lysate by centrifugation for 10 min (4°C, 800 × g), treated with 0.2% (v/v) Triton X-100 (10 min, 37°C) and washed twice with sodium phosphate buffer. Cell debris were removed from the cell lysate by additional centrifugation at high speed (10 min, 4°C, 16,000 × g). Soluble and inclusion body fractions were analysed by SDS-PAGE and Western blotting.
Coexpression of the BVMO gene with chaperone encoding plasmids
E. coli JM109 and BL21 (DE3) cells were transformed with chaperone encoding plasmids and competent cells were prepared thereof. These were transformed with pJOE constructs (for JM109) or pET22b(+) constructs (for BL21 (DE3)) and selected on LB-plates containing 100 μg ampicillin/ml and 34 μg chloramphenicol/ml (LBamp + cm). Expression was performed at 30°C as described before using LBamp + cm containing 0.5 mg l-arabinose/ml (in case of pGro7, pKJE7 and pTf16), 5 ng tetracycline/ml (in case of pG-Tf2) or l-arabinose and tetracycline (in case of pG-KJE8) in concentrations given above.
Biocatalysis and biotransformation
Substrate specificity was investigated using crude cell extract, growing and resting cells. For all experiments E. coli JM109 pJOE-KT2440 pGro7 cells were used.
Biocatalysis with crude cell extract
Cells were grown in 2x YTamp + cm medium at 37°C to an optical density (600 nm) of 0.6–0.7. BVMO expression was induced by the addition of l-rhamnose to 0.2% (w/v), chaperone expression (GroES/EL) by the addition of l-arabinose to 0.5 mg/ml. Expression was performed at 25°C overnight (∼12 h). Cells were harvested at an optical density (600 nm) of 6–7, washed and resuspended in sterile sodium phosphate buffer (50 mM, pH 7.5). Cell disruption was performed by sonication on ice, cell debris were removed by centrifugation (30 min, 4°C, 10,000 × g). Biocatalysis reactions were performed in 2 ml reaction tubes closed with air permeable caps (LidBac, Eppendorf) at 20, 25 and 30°C. For all investigated temperatures the same crude cell extract was used. To 900 μl crude cell lysate 50 μl FAD (1 mg/ml), 200 mM NADPH and 50 mM substrate (aliphatic, aromatic and cyclic ketones) were added. After defined time intervals (1, 2, 4 and 6 h) samples were taken, extracted with dichloromethane and dried over anhydrous sodium sulphate. Samples were analysed via GC-MS.
Biotransformation with resting cells
Cultivation and BVMO expression were performed as described for crude cell extract. At an optical density (600 nm) of 6–7, cells were harvested, washed twice with sterile sodium phosphate buffer (50 mM, pH 7.5) and resuspended in the same buffer. 10 mM substrate and 20 mM glucose for cofactor regeneration were added simultaneously. Biotransformation was performed in flasks (50 ml cell suspension; optical density 6–7) at 20, 25 and 30°C. Samples were taken at defined times (0.5, 1, 2, 4, 8, 24 and 32 h) and treated as above.
Biotransformation with growing cells
Cells were grown in 50 ml 2x YTamp + cm at 37°C to an optical density at 600 nm of 0.6–0.7. BVMO expression was induced by addition of l-rhamnose to 0.2% (w/v) and chaperone expression (GroES/EL) by the addition of l-arabinose to 0.5 mg/ml. Subsequently, 10 mM substrate was added. Expression and biotransformation were performed at 20, 25 and 30°C. After defined time intervals (1, 4, 8 and 24 h) samples were taken and treated as described above.
GC-MS analysis
GC-MS analyses were carried out on a GCMS-QP 2010 instrument (Shimadzu) with a BPX5 column (Macherey-Nagel). All substrates, methyl and ethyl esters were purchased from Fluka at GC standard purity. Other ester standards for GC-MS analysis were prepared enzymatically according to Kirschner et al. (2007). 1H-NMR spectra were recorded in CDCl3 on a 300 MHz (Bruker) instrument. The obtained NMR spectra of aliphatic esters matched literature data.
Results and discussion
Cloning and BVMO expression in E. coli
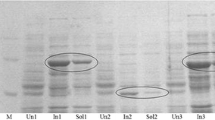
The gene of the BVMO from Pseudomonas putida KT2440 was successfully amplified by PCR from the genomic DNA and inserted into pJOE4072.6 and pET22b(+). The BVMO expression was performed in E. coli JM109 with pJOE-KT2440 and in BL21 (DE3) with pET22b(+)-KT2440. Different expression temperatures were investigated and the amount of BVMO in the soluble and inclusion body fraction was studied by SDS-PAGE and western blotting. For both systems, more enzyme was found in the pellet than in the soluble fraction revealing a problem with the proper folding of the BVMO in E. coli. Therefore, the BVMO was co-expressed at 30°C with different combinations of chaperones provided by the TaKaRa Chaperone Plasmid Set. It has already been reported that coexpression of molecular chaperones in the E. coli cytoplasm could significantly increase the yield of soluble protein (Lee et al. 2004). For co-expression in E. coli JM109, the GroES/GroEL complex (pGro7) as well as GroES/GroEL together with DnaK/DnaJ/GrpE (pG-KJE8) or the trigger factor (pG-Tf2) led to noticeably higher amounts of soluble BVMO (Fig. 1), whereas the DnaK/DnaJ/GrpE (pKJE7) alone or in combination with the trigger factor (pTf16) showed only a slightly positive effect on BVMO expression (data not shown). For coexpression in BL21 (DE3) no noticeably higher amount of soluble BVMO could be obtained (data not shown).

SDS-PAGE and Western blot of the soluble fraction of BVMO expression in E. coli JM109 with and without coexpression of chaperones [a]; for detection of 6xHis tagged BVMO, the QIAexpress detection system using a Ni-NTA-alkaline phosphatase conjugate (Qiagen) was applied according to the manufacturer’s instructions. Notes: [a] Lanes 1–3: expression without chaperones at 25, 30 and 37°C, respectively; lanes 5–7: chaperone coexpression with pG-KJE8, pGro7 and pG-Tf2 at 25°C, respectively; lane 4: (a) RotiMark Standard (Roth), (b) Low Range Marker (Sigma). [b] Western blot analysis of expression experiments with E. coli JM109 pJOE without an additional gene revealed a band of the same molecular weight assuming an E. coli host-protein with cross-reaction to the Ni-NTA-alkaline phosphatase conjugate (data not shown)
Substrate specificity
In order to investigate the substrate specificity of the BVMO from Ps. putida KT2440 biocatalytic reactions at 30°C using crude cell extract were performed. Different substrates (cyclic, aromatic and aliphatic open-chain ketones) were investigated. However, only aliphatic acyclic ketones were converted to the corresponding esters with good conversions, while oxidation of cyclic ketones (cyclohexanone, cyclopentanone and cycloheptanone) was rather low and aromatic ketones (acetophenone and derivatives) were not accepted at all (data not shown). In fact, a search in the protein sequence data base revealed that one of the closest homologs (52% sequence identity) with known substrate specificity is the prodrug activator EtaA from Mycobacterium tuberculosis (Fraaije et al. 2004). This enzyme was shown to convert aliphatic open-chain ketones as well as phenylacetone and benzylacetone, while typical BVMO substrates like acetophenone and cyclohexanone were not accepted. Interestingly, sequence identity with a BVMO from Ps. fluorescens DSM50106 with similar substrate specificity was only 25% (Kirschner et al. 2007).
Further experiments investigating the conversion of aliphatic open-chain ketones (C8–C13) were then performed at 20, 25 and 30°C using crude cell extract, growing and resting cells. When using whole cells expressing the BVMO, there was no need to add NADPH, which makes biotransformations cost-effective. Reaction courses were monitored via GC-MS analysis.
In Table 1, conversions at 25°C after 8 h reaction time for all investigated aliphatic acyclic ketones using the three different systems are summarized. As a control, biotransformations were also performed under the same reaction conditions using a pJOE-vector without the BVMO gene, but no ester formation could be detected. Additionally, no other by-product could be found and product esters were not further hydrolyzed by E. coli enzymes since no alcohol could be detected. However, especially for the shorter chain compounds (C8, C9) a significant loss of substrate and product due to evaporation was evident at 30°C.
Apparently, in all three systems the BVMO from Ps. putida KT2440 preferentially converted short-chain ketones like 2-decanone, 3-decanone and 4-decanone. Here conversions reached the highest values. A BVMO from Ps. cepacia also converts aliphatic open-chain ketones with good yields. However, in contrast to the BVMO from Ps. putida KT2440, this 2-tridecanone monooxygenase preferred medium-chain ketones from C12–C14 and also converted cyclopentanone with high specific activity (Britton and Markovetz 1977).
Comparing biocatalysis with crude cell extract and biotransformation using whole cells, either growing or resting, revealed that the latter system worked more efficiently at all investigated temperatures since conversions were higher and some ketones were oxidized even quantitatively. In the case of crude cell extract, sonication leading to the loss of the enzyme protecting physiological environment can influence enzyme activity and stability. This might be a possible explanation for reduced conversions. When using growing cells, substrate was added simultaneously to the time-point of induction of BVMO expression, whereas for resting cells the enzyme was first expressed before starting biotransformations. Conclusively, the reaction times for growing cells are much longer compared to resting cells. Another problem using growing cells was that 2-nonanone (or an unknown impurity in the substrate) seemed to have a harmful effect, since no conversion could be detected and no more growth could be observed after substrate addition. Thus, resting cells seemed to give best results working sufficiently and cost-effective.
Interestingly, conversion of some ketones did not only generate one ester product. According to Fig. 2, oxygen can be inserted on both sides of the keto group producing alkylacetates as well as the less favored methyl esters. With longer chain length the enzyme does not insert oxygen by migration of the higher substituted alkyl chain as typical for Baeyer-Villiger reactions. Instead, conversion into the methylesters seem to be preferred. Comparing these results to those of the BVMO from Ps. fluorescens DSM50106 it strikes that for this enzyme the migrating substituent during oxygen insertion depends on the position of the keto group. While here all 2-ketones became acetates, 3-ketones were converted into both ester products and 4-ketones gave propylesters only (unpublished results). In the case of EtaA from Mycobacterium tuberculosis insertion of oxygen depends on alkyl chain length and position of the keto group (Fraaije et al. 2004).

BVMO catalyzed conversion of aliphatic open-chain ketones
Examining the different results at 20, 25 and 30°C also revealed a correlation between reaction temperature and conversion. Biotransformations at 30°C using resting cells lead to a two-fold increase or quantitative conversion in comparison to 20°C after the same reaction time (data not shown). At higher temperatures reaction times were shorter but, especially for the short-chain ketones, the effect of evaporation was also more evident.
Conclusion
In this study, the gene of a Baeyer-Villiger monooxygenase from Pseudomonas putida KT2440 was successfully amplified from genomic DNA and cloned into two different vectors. Performing coexpression of different chaperones with the BVMO successfully lead to soluble protein. Investigation of substrate specificity revealed a high preference towards short-chain aliphatic ketones. Comparing biocatalysis using crude cell extract and whole cells showed that resting cells worked most efficiently and gave the best results. Additionally, it was found that some acyclic ketones were converted not only to the alkylacetates, but also methyl- and ethylesters were obtained, indicating insertion of oxygen on both sides of the keto group.
References
Britton LN, Markovetz AJ (1977) A novel ketone monooxygenase from Pseudomonas cepacia. J Biol Chem 252:8561–8566
Donoghue NA, Norris DB, Trudgill PW (1976) The purification and properties of cyclohexanone oxygenase from Nocardia globerula CL1 and Acinetobacter calcoaceticus NCIMB 9871. Eur J Biochem 63:175–192
Fraaije MW, Kamerbeek NM, Heidekamp AJ et al (2004) The produg activator EtaA from Mycobacterium tuberculosis is a Baeyer-Villiger monooxygenase. J Biol Chem 279:3354–3360
Fraaije MW, Kamerbeek NM, van Berkel WJH et al (2002) Identification of a Baeyer-Villiger monooxygenase sequence motif. FEBS Lett 518:43–47
Kamerbeek NM, Moonen MJH, van der Ven JGM et al (2001) 4-Hydroxyacetophenone monooxygenase from Pseudomonas fluorescens ACB. Eur J Biochem 268:2547–2557
Kirschner A, Altenbuchner J, Bornscheuer UT (2007) Cloning, expression and characterization of a Baeyer-Villiger monooxygenase from Pseudomonas fluorescens DSM 50106 in E. coli. Appl Microbiol Biotechnol 73:1065–1072
Kirschner A, Bornscheuer UT (2006) Baeyer-Villiger monooxygenase-catalyzed kinetic resolution of 4-hydroxy-2-ketones. Angew Chem Int Ed 45:7004–7006
Lee DH, Kim MD, Lee WH et al (2004) Consortium of fold-catalyzing proteins increases soluble expression of cyclohexanone monooxygenase in recombinant Escherichia coli. Appl Microbiol Biotechnol 63:549–552
Malito E, Alfieri A, Fraaije MW et al (2004) Crystal structure of a Baeyer-Villiger monooxygenase. Proc Natl Acad Sci USA 101:13157–13162
Mihovilovic MD, Müller B, Stanetty P (2002) Monooxygenase-mediated Baeyer-Villiger oxidations. Eur J Org Chem 2002:3711–3730
Nelson KE, Weinel C, Paulsen IT et al (2002) Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ Microbiol 4:799–808
Stewart JD (1998) Cyclohexanone monooxygenase: a useful reagent for asymmetric Baeyer-Villiger reactions. Curr Org Chem 2:211–232
Tanner A, Hopper DJ (2000) Conversion of 4-hydroxyacetophenone into 4-phenyl acetate by a flavin adenine dinucleotide-containing Baeyer-Villiger-type monooxygenase. J Bacteriol 182:6565–6569
Acknowledgements
We thank the Deutsche Bundesstiftung Umwelt (Osnabrück, Germany) for a stipend to Jessica Rehdorf and the Fonds der Chemischen Industrie (Frankfurt, Germany) and the Studienstiftung des Deutschen Volkes (Bonn, Germany) for stipends to Anett Kirschner.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rehdorf, J., Kirschner, A. & Bornscheuer, U.T. Cloning, expression and characterization of a Baeyer-Villiger monooxygenase from Pseudomonas putida KT2440. Biotechnol Lett 29, 1393–1398 (2007). https://doi.org/10.1007/s10529-007-9401-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-007-9401-y