
Abstract
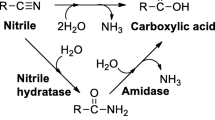
2,6-Pyridinedicarbonitrile (1a) and 2,4-pyridinedicarbonitrile (2a) were hydrated by Rhodococcus erythropolis A4 to 6-cyanopyridine-2-carboxamide (1b; 83% yield) and 2-cyanopyridine-4-carboxamide (2b; 97% yield), respectively, after 10 min. After 118 h, the intermediates 1b or 2b were transformed into 2,6-pyridinedicarboxamide (1c; 35% yield) and 2,6-pyridinedicarboxylic acid (1d; 60% yield) or 2-cyanopyridine-4-carboxylic acid (2c; 64% yield), respectively. The nitrilase from Fusarium solani afforded cyanocarboxylic acids 1e and 2c after 118 h (yields 95 and 62%, respectively). 3,4-Pyridinedicarbonitrile (3a) and 2,3-pyrazinedicarbonitrile (4a) were inferior substrates of nitrile hydratase and nitrilase.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Previously, we reported on biocatalysts suitable for the mild hydrolysis of heterocyclic nitriles. A nitrilase from Aspergillus niger exhibited high activities for 3- and 4-cyanopyridine (Kaplan et al. 2006a). The latter compound was hydrolyzed at the highest rate of all substrates tested, probably due to an electron withdrawing effect of the heteroatom. Different immobilized formulations of the nitrilase from Fusarium solani were also highly active towards the above nitriles (Vejvoda et al. 2006a). In addition, a nitrile hydratase from Rhodococcus erythropolis A4 (formerly R. equi A4) was useful for the preparation of amides from heterocyclic nitriles like cyanopyridines (Přepechalová et al. 2001).
Considering the potential of these enzymes for the production of heterocyclic acids or amides, we became interested in their activity towards heterocyclic dinitriles. Transformation of aliphatic and aromatic dinitriles attracted much attention due to its utility for the preparation of useful compounds bearing diverse functional groups (for reviews see Sugai et al. 1997 and Martínková and Mylerová 2003). In this work, we examined the feasibility of using nitrilase and nitrile hydratase for the transformations of 2,6-, 2,4- and 3,4-pyridinedicarbonitrile and 2,3-pyrazinedicarbonitrile.
Materials and methods
Chemicals
The substrates (see Fig. 1) were 2,6-pyridinedicarbonitrile (1a), 2,4-pyridinedicarbonitrile (2a), 3,4-pyridinedicarbonitrile (3a), 2,3-pyrazinedicarbonitrile (4a) and benzonitrile. The authentic standards (see Fig. 2) were 2,6-pyridinedicarboxamide (1c) and 2,6-pyridinedicarboxylic acid (1d). All substrates and standards were purchased from Aldrich.

Heterocyclic dinitriles used as substrates of nitrilases and nitrile hydratase/amidase

Products formed from 2,6-pyridinedicarbonitrile (1a) by whole cells of Rhodococcus erythropolis (nitrile hydratase/amidase; i) and cell-free extract from Fusarium solani (nitrilase; ii)
HPLC
Nitriles, amides and acids were analyzed by HPLC with a Novapak C18 column (4 μm, 150 × 3.9 mm; Waters). Benzonitrile, benzamide and benzoic acid were separated using a mobile phase composed of acetonitrile:water:phosporic acid (30:69.9:0.1) at a flow rate of 0.9 mol min−1 and 35°C. For separation conditions, retention times and spectral maxima of compounds 1a–1e, 2a–2c, 3a and 4a see Table 1.
Preparative HPLC was performed with a Nucleosil 120–5 C18 column (5 μm, 250 × 8 mm; Macherey-Nagel) at of 0.5 ml min−1 and ambient temperature. The mobile phase was composed of acetonitrile:water:acetic acid (10:89:1, by vol.).
16S rDNA sequencing
A fragment of 16S rDNA (881 bp) was amplified by PCR using the total genome DNA of the Rhodococcus strain A4 as a template and oligonucleotides 16SREF and 16SRER (Čejková et al. 2005) as primers. The sequence of the fragment was determined with ABI Prism 2100 sequencer (PerkinElmer). The sequence was found identical (100%) with the respective part of the homologous sequence of R. erythropolis 16S rDNA stored in the GenBank while there was only 96% identity with R. rhodochrous 16S rDNA and 95% identity with R. equi 16S rDNA. The strain A4, originally classified as R. equi according to biochemical tests, was thus reclassified as R. erythropolis on the basis of the similarity searches of 16S rDNA sequences.
Biocatalysts
Whole cells of Rhodococcus erythropolis A4 (deposited in the Culture Collection of Microorganisms, Masaryk University Brno) were grown as described by Martínková et al. (1998). The nitrile hydratase was partially purified as described by Přepechalová et al. (2001). The nitrilases from Aspergillus niger CCF 3411 (Culture Collection of Fungi, Charles University Prague, Czech Republic) and Fusarium solani CCF 3635 were used as a semi-purified enzyme (Kaplan et al. 2006a) and a cell-free extract (Vejvoda et al. 2006a), respectively. The enzyme assays are described below. The general procedure for preparative-scale biotransformations is described in Table 3.
Enzyme assays
Nitrile hydratase and nitrilase activities were assayed in 2 ml reaction mixtures composed 20 μmol benzonitrile in 50 mM Na2HPO4/KH2PO4 buffer, pH 8, and 5% (v/v) methanol as co-solvent. The reactions were started by the addition of an appropriate amount of the enzyme (approx. 0.2 U as assayed for benzonitrile). The nitrile hydratase- and nitrilase-catalyzed reactions proceeded at 32°C and 45°C, respectively, with shaking (850 rpm). After 10 min, the reactions were stopped by the addition of 20% (v/v) of 1 M HCl and the precipitated protein was removed by centrifugation. Benzamide and benzoic acid were determined by HPLC. Activities of the enzymes towards substrates 1a–4a were assayed in the same way and substrate consumption was determined by HPLC. The product formation and substrate consumption was linear within the reaction time.
Product identification
Using authentic standards, products 1c and 1d were identified by HPLC and by UV spectra recorded by the PDA detector. Other products (1b, 1e, 2b and 2c) were identified by NMR spectra recorded on a 400 MHz spectrometer (Varian unity Inova-400) in DMSO-d6 at 30°C. The assignment was based on COSY, HMQC and HMBC experiments performed using the manufacturer’s software. Residual solvent signals (DMSO: δH 2.50 ppm, δC 39.60 ppm) were used as internal standards.
Results and discussion
Substrate specificity of nitrile-converting enzymes towards heterocyclic dinitriles
Pyridine derivatives with cyano groups in meta-position (1a, 2a) were good substrates of the nitrile hydratase from R. erythropolis (see Table 2). Steric hindrance by heteroatom probably decreased the reactivity of compound 1a, which was transformed at approx. 3 times lower rate than compound 2a. Not suprisingly, the activities of the nitrile hydratase towards dinitriles with cyano groups in ortho-position (3a, 4a) were even lower. This enzyme was previously shown to be influenced by steric effects of ortho-substituents (Přepechalová et al. 2001).
The nitrilase from F. solani is more susceptible towards steric hindrances than that from A. niger (Kaplan et al. 2006b) and this observation is in accordance with lower activities of the former enzyme towards compounds 1a and 2a (see Table 2). This nitrilase showed no activity towards ortho-substituted compounds 3a and 4a, while the nitrilase from A. niger was able to hydrolyze them at a very low rate.
Products of enzymatic hydrolysis of heterocyclic dinitriles
The above experiments suggested the feasibility of preparative biotransformations of dinitriles 1a and 2a by both nitrile hydratase and nitrilase. Previously, we observed that both enzymes hydrolyzed a single cyano group in dicyanobenzenes (Přepechalová et al. 2001, Kaplan et al. 2006a, b). Therefore, we studied the mode of action of these enzymes towards pyridinedicarbonitriles. Whole cells of R. erythropolis were used as the source of nitrile hydratase in preparative biotransformations. This strain also exhibited amidase activity. A cell-free extract from F. solani was used for the preparative nitrilase-catalyzed biotransformations despite its lower specific activity for 1a and 2a, as this enzyme was available in large quantities from hyperinduced cultures (Vejvoda et al. 2006a). In addition, the nitrilase from A. niger often produced large amounts of by-products-amides (Kaplan et al. 2006a) and, therefore, seemed to be less suitable for preparative biotransformation than the enzyme from F. solani, which gave nearly pure carboxylic acids (Vejvoda et al. 2006a).
The biotransformations of 1a and 2a were monitored by HPLC within 0–5 days. Compound 1a was transformed into three major products by whole cells of R. erythropolis, while compound 2a gave only two major products on incubation with this biocatalyst. The nitrilase from F. solani gave one major product from each substrate. The products were isolated and identified during the reaction (see Table 3). In general, purification by preparative HPLC was necessary due to cross-contamination of the products and unreacted nitriles. The isolated yields were not optimized in this procedure, the aim of which was to obtain pure biotransformation products for structure elucidation. Commercial authentic standards were available only for diamide 1c and diacid 1d. The reaction patterns for substrates 1a and 2a are shown in Figs. 2 and 3, respectively. Analytical yields of the products at different reaction times are given in Table 3.

Products formed from 2,4-pyridinedicarbonitrile (2a) by whole cells of Rhodococcus erythropolis (nitrile hydratase/amidase; i) and cell-free extract from Fusarium solani (nitrilase; ii)
The biotransformation of substrate 1a catalyzed by nitrile hydratase gave cyano amide 1b after 10 min. This intermediate was then hydrated into diamide 1c within approx. 2 h. The following amidase-catalyzed reaction was slow. The end product of this reaction was dicarboxylic acid 1d, which was the major product after 118 h. On the other hand, the reaction of 2a showed a different course. The corresponding cyano amide 2b was also rapidly formed at the initial stage (10 min) but afterwards slowly transformed into cyano carboxylic acid 2c as the major product by the amidase (after 118 h).
The products of nitrilase-catalyzed reactions were identified as cyanocarboxylic acids 1e and 2c. The reactions of 2a catalyzed by both nitrile hydratase and nitrilase were regioselective, only the nitrile group distant from the heteroatom being susceptible to hydration or hydrolysis.
Formation of amides as by-products by nitrilases
The production of amides by nitrilases is probably caused by an atypical cleavage of the reaction intermediate (Stevenson et al. 1992). It was especially pronounced for some substrates with electron-withdrawing groups (Effenberger and Osswald 2001, Kiziak et al. 2005). Nevertheless, this phenomenon is also strongly strain-dependent. The nitrilase from F. solani (Vejvoda et al. 2006a) formed no or only low amounts of amides from nitriles, as also observed in the present work. On the other hand, the nitrilase from A. niger (Kaplan et al. 2006a) formed high amounts of amides, especially from substrates with an electron-withdrawing heteroatom (2-cyanopyridine, 4-cyanopyridine). The substrates examined herein also contained electron withdrawing moieties (nitrogen heteroatom, cyano groups). Therefore, we examined the products formed by A. niger nitrilase from compounds 1a and 2a. Indeed, the corresponding amide was formed in both cases. The molar acid:amide ratio (1c:1b––9:1, 2c:2b––7:3) was constant during the reaction. This side reaction may complicate the biocatalytical use of nitrilase from A. niger. On the other hand, the benefit of this enzyme is its higher specific activity towards sterically hindered substituted nitriles like 1a and 2a. Recently, we proposed to use a bienzymatic (nitrilase-amidase) reaction to remove the by-product amide (Vejvoda et al. 2006b).
Conclusion
The enzymatic hydration or hydrolysis of 2,6-pyridinedicarbonitrile and 2,4-pyridinedicarbonitrile provided access to heterocyclic compounds bearing multiple functional groups in their molecules. These compounds can serve as useful intermediates for chemical synthesis. The product type (cyano amide, cyano acid) could be controlled by the choice of an appropriate enzyme and by the reaction time.
References
Čejková A, Masák J, Jirků V, Veselý M, Pátek M, Nešvera J (2005) Potential of Rhodococcus erythropolis as a bioremediation organism. World J Microbiol Biotechnol 21:317–321
Effenberger F, Osswald S (2001) Enantioselective hydrolysis of (RS)-2-fluoroarylacetonitriles using nitrilase from Arabidopsis thaliana. Tetrahedron: Asymmetry 12:279–285
Kaplan O, Nikolaou K, Pišvejcová A, Martínková L (2006) Hydrolysis of nitriles and amides by filamentous fungi. Enzyme Microb Technol 38:260–264
Kaplan O, Vejvoda V, Plíhal O, Pompach P, Kavan D, Bojarová P, Bezouška K, Macková M, Cantarella M, Jirků V, Křen V, Martínková L (2006) Purification and characterization of a nitrilase from Aspergillus niger K10. J Appl Microbiol Biotechnol 73:567–575
Kiziak C, Conradt D, Stolz A, Mattes R, Klein J (2005) Nitrilase from Pseudomonas fluorescences EBC 191: cloning and heterologous expression of the gene and biochemical characterization of the recombinant enzyme. Microbiology 151:3639–3648
Martínková L, Klempier N, Přepechalová I, Přikrylová V, Ovesná M, Griengl H, Křen V (1998) Chemoselective biotransformation of nitriles by Rhodococcus equi A4. Biotechnol Lett 20:909–912
Martínková L, Mylerová V (2003) Synthetic applications of nitrile-converting enzymes. Curr Org Chem 7:1279–1295
Přepechalová I, Martínková L, Stolz A, Ovesná M, Bezouška K, Kopecký J, Křen V (2001) Purification and characterization of the enantioselective nitrile hydratase from Rhodoccoccus equi A4. Appl Microbiol Biotechnol 55:150–156
Stevenson DE, Feng R, Dumas F, Groleau D, Mihoc A, Storer AC (1992) Mechanistic and structural studies on Rhodococcus ATCC 39484 nitrilase. Biotechnol Appl Biochem 15:283–302
Sugai T, Yamazaki T, Yokoyama M, Ohta H (1997) Biocatalysis in organic synthesis: the use of nitrile- and amide-hydrolyzing microorganisms. Biosci Biotech Biochem 61: 1419–1427
Vejvoda V, Kaplan O, Klozová J, Masák J, Čejková A, Jirků V, Stloukal R, Martínková L (2006) Mild hydrolysis of nitriles by Fusarium solani strain O1. Folia Microbiol 51:251–256
Vejvoda V, Kaplan O, Kubáč O, Křen V, Martínková L (2006) Immobilization of fungal nitrilase and bacterial amidase – two enzymes working in accord. Biocatal Biotrans 24:414–418
Acknowledgement
Financial support through projects IAA500200708 (Grant Agency of the Academy of Sciences of the Czech Republic), LC06010 and OC D25.001 (Ministry of Education, Czech Republic) and the institutional research concept AV0Z50200510 (Institute of Microbiology) is gratefully acknowledged. We also wish to thank Mrs. Jana Horová for her excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
6-Cyanopyridine-2-carboxamide (1b) 1H NMR (399.87 MHz, DMSO-d 6 , 30°C): 7.830 (1 H, br s, NH2-u), 8.211 (1H, dd, J = 7.6, 2.2 Hz, H-5), 8.22 (1H, br s, NH2-d), 8.238 (1 H, dd, J = 7.6, 7.0 Hz, H-4), 8.294 (1H, dd, J = 7.0, 2.2 Hz, H-3). 13C NMR (100.55 MHz, DMSO-d 6 , 30°C): 117.09 (6-CN), 125.88 (C-3), 131.31 (C-5), 131.46 (C-6), 139.75 (C-4), 151.91 (C-2), 164.49 (2-CO).
6-Cyanopyridine-2-carboxylic acid (1e) 1H NMR (399.87 MHz, DMSO-d 6 , 30°C): 8.231 (2 H, m, H-4, H-5), 8.296 (1 H, m, H-3). 13C NMR (100.55 MHz, DMSO-d 6 , 30°C): 117.01 (6-CN), 128.21 (C-3), 131.64 (C-5), 132.57 (C-6), 139.63 (C-4), 150.23 (C-2), 164.77 (2-CO).
2-Cyanopyridine-4-carboxamide (2b) 1H NMR (399.87 MHz, DMSO-d 6 , 30°C): 7.948 (1 H, br s, NH), 8.078 (1 H, dd, J = 5.1, 1.7 Hz, H-5), 8.347 (1 H, dd, J = 1.7, 0.9 Hz, H-3), 8.370 (1 H, br s, NH), 8.904 (1 H, dd, J = 5.1, 0.9 Hz, H-6). 13C NMR (100.55 MHz, DMSO-d 6 , 30°C): 117.29 (2-CN), 125.49 (C-5), 126.69 (C-3), 133.26 (C-2), 142.92 (C-4), 152.11 (C-6), 164.55 (4-CO).
2-Cyanopyridine-4-carboxylic acid (2c) 1H NMR (399.87 MHz, DMSO-d 6 , 30°C): 8.079 (1 H, dd, J = 4.9, 1.3 Hz, H-5), 8.296 (1 H, s, H-3), 8.872 (1 H, d, J = 4.9 Hz, H-6). 13C NMR (100.55 MHz, DMSO-d 6 , 30°C): 117.37 (2-CN), 126.85 (C-5), 128.05 (C-3), 133.25 (C-2), 143.11 (C-4), 152.07 (C-6), 164.82 (4-CO).
Rights and permissions
About this article
Cite this article
Vejvoda, V., Šveda, O., Kaplan, O. et al. Biotransformation of heterocyclic dinitriles by Rhodococcus erythropolis and fungal nitrilases. Biotechnol Lett 29, 1119–1124 (2007). https://doi.org/10.1007/s10529-007-9364-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-007-9364-z