
Abstract
Inter-retrotransposon amplified polymorphisms (IRAPs) and retrotransposon-microsatellite amplified polymorphisms (REMAPs) were used to detect retrotransposon integration events and genetic diversity in 101 Iranian bread wheat (Triticum aestivum L.) cultivars and breeding lines. The 9 IRAP primers amplified 128 loci, and 20 REMAP primers amplified 263 loci. Percentage of polymorphic loci, average expected heterozygosity, number of effective alleles, and Shannon’s information index for the REMAP markers were slightly higher than those for the IRAP markers. The same estimated parameters calculated for native and nonnative retrotransposons were not considerably different. A Mantel test between IRAP and REMAP cophenetic matrices evidenced no significant correlation. Cluster analysis based on the Dice similarity coefficient and complete linkage algorithm using IRAP+REMAP loci identified five groups among the genotypes studied that could be applied as crossing parents in T. aestivum breeding programs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bread wheat (Triticum aestivum L.), as a widely grown crop in many countries including Iran, was domesticated 12,000 years ago in the Fertile Crescent (Salamini et al. 2002). In the cradle of agriculture, Iran is considered a center for the domestication of wheat and of its genetic diversity. The available genetic variation was subsequently reduced by genetic drift and selection in the wild and by early farmers. A decrease in allelic diversity after the 1960s was demonstrated in a study of 559 French bread wheat accessions from the years 1800–2000 using 42 SSRs (Roussel et al. 2004) and in 480 European wheat cultivars from 1840–2000 using 39 SSRs (Roussel et al. 2005). In Iran, wheat breeding commenced in 1942, and new cultivars were developed from local populations and a set of genetically related, modern high-yielding, introduced varieties. As a result, many landraces were continuously replaced by modern wheat cultivars. Therefore, Iranian wheat germplasm has experienced an overall reduction of its genetic basis as a result of high selection pressure, recurrent use of the adapted elite germplasm, and the adoption of breeding schemes that have not favored genetic recombination (Mohammadi et al. 2009; Saeidi et al. 2008). Such reduction may have serious consequences, as seen in the epidemic extension of yellow rust in Iran in 2009.
In recent decades, in collaboration with international research centers such as CIMMYT, a number of new genotypes and breeding lines have been introduced to the Iranian national wheat breeding programs to increase the genetic variation of its wheat germplasm. Similar to most germplasms, only a small proportion of the available genetic variation has been exploited for plant breeding in wheat and other crop plants (Tanksley and McCouch 1997). Most of the germplasm remains untapped, uncharacterized, and unutilized (Fernie et al. 2006). Therefore, the genetic variation provided by the current and expanded gene pools should be characterized for further utilization in crop improvement programs.
Retrotransposons (RTNs) have been detected in most grasses, accounting for as much as 80% of the genome (San Miguel et al. 1996). The RTNs replicate by successive transcription, reverse transcription, and insertion of new 5–10 kb cDNA copies back into the genome as well as the retroviruses themselves, increasing the genome size (Kumar and Bennetzen 1999). The dispersion, ubiquity, and prevalence of RTNs in plant genomes allowed their success as DNA molecular marker systems based on the PCR technique. The new insertions promoted by replicationally active members of an RTN family result in insertional polymorphisms that can be detected by inter-retrotransposon amplified polymorphism (IRAP) and retrotransposon-microsatellite amplified polymorphism (REMAP) (Kalendar et al. 1999). IRAP has been used alone or combined with REMAP in studies of genetic diversity and phylogeny within several plant genera and species, including Hordeum (Kalendar et al. 1999), Citrus (Biswas et al. 2010), Pisum (Smykal 2006), Oryza (Branco et al. 2007), Aegilops (Saeidi et al. 2008), and Medicago sativa L. (Abdollahi Mandoulakani et al. 2012). Direct comparisons of RTN methods with other marker systems indicate that the RTN markers are more informative and polymorphic in a variety of crops (Queen et al. 2004; Tam et al. 2005; Abdollahi Mandoulakani et al. 2012). RTN marker data are more consistent with geographical and morphological criteria than amplified fragment length polymorphism (AFLP) markers (Ellis et al. 1998). A major disadvantage of RTN-based molecular markers is the need for RTN sequence information to design family-specific primers. However, related species have similar RTN sequences, meaning that primers designed from long terminal repeat (LTR) sequences of an RTN can be readily used across species lines, among closely related genera, and even sometimes between plant families (Lou and Chen 2007; Kalendar et al. 2011).
In the present study, we used IRAP and REMAP markers to detect integration events and activity of LTR RTN families isolated from wheat and barley in the Iranian bread wheat genome. IRAP and REMAP markers derived from these RTNs were also used to assess the genetic variability among 101 Iranian bread wheat cultivars and breeding lines in order to characterize this interesting germplasm, which needs to be conserved, maintained, and further used in breeding programs to widen the genetic basis of modern wheat varieties and to avoid the loss of rare and unique alleles.
Materials and Methods
Plant Material and DNA Extraction
Plant material consisted of 50 Iranian bread wheat (Triticum aestivum L.) cultivars and 51 breeding lines (Supplementary Table 1) kindly provided by the Seed and Plant Improvement Institute, Karaj, Iran. Genomic DNA was extracted from young leaves of 20-day seedlings of 5 plants from each genotype using the method described by Ausubel et al. (1995) with minor modifications. The quality and concentration of the DNA were measured using a spectrophotometer and electrophoresis in a 0.8% (w/v) agarose gel.
IRAP and REMAP Reactions
Eleven single and 30 IRAP primer combinations (Supplementary Table 2; Table 1) were tested on 6 cultivars and lines to select the primers producing discernible and distinguishable banding patterns. The RTNs WIS2-1A, Wis, Wilma, Daniela, and Fatima, originally identified from wheat, were considered native RTNs. Bare1 and Sukkula identified in barley were considered nonnative RTNs. The IRAP and REMAP primers were divided into native and nonnatives to compare their polymorphism level and diversity. IRAP primer combinations with one nonnative RTN were defined as nonnative primers. In REMAP, primer combinations with native RTNs were considered native primers, and the rest were considered nonnatives. Based on these criteria, 20 primers were defined as native and 9 as nonnative.
IRAP PCR amplifications were carried out in a Bio-Rad thermo cycler (Bio-Rad Laboratories, Hercules, CA, USA) in a total volume of 20 μl containing 45 ng genomic DNA, 1× PCR buffer (10 mM Tris–HCl, 50 mM KCl, pH 8.3), 1.5 mM MgCl2, 0.2 μM of each dNTP, 0.5 U Taq DNA polymerase (Cinagen, Iran), and 10 pmol of each primer. The amplification profile consisted of an initial denaturation at 94°C for 4 min, followed by 36 cycles at 94°C for 40 s, 57–63°C (Table 1) for 40 s, 72°C for 2 min, and a final extension at 72°C for 5 min. PCR products were separated by electrophoresis (Bio-Rad) using 1.8% Resolute Line Biozyme agarose gel in 0.5× TBE buffer with a constant voltage of 65 V for 3–4 h. Gels were stained by ethidium bromide. DNA fragments were then visualized under UV light and photographed using a gel documentation system.
There were 88 REMAP primer combinations, derived from 11 single IRAP primers with 8 ISSR primers (Supplementary Table 2; Table 1). They were tested on 6 bread wheat genotypes to select the primer combinations generating scorable and discernible banding patterns. PCR amplification reactions and temperature profile, electrophoresis, and visualization of REMAP markers were as for IRAP. Annealing temperatures of IRAP and REMAP primer combinations were optimized by gradient PCR and depended on the primers (Table 1).
Statistical Analyses
The amplified fragments were scored independently as 1 (present) or 0 (absent) at each position, and the binary data obtained were used for analyses. Three genetic similarity matrices, for IRAP, REMAP, and combined data (IRAP+REMAP), were calculated among 101 genotypes using the Dice similarity coefficient and were subsequently used to construct three dendrograms based on the complete linkage algorithm. To verify the adjustment between similarity matrices and respective dendrogram-derived matrices (cophenetic matrix), the cophenetic correlation coefficient was estimated. To estimate the degree of correlation among the three cophenetic matrices for IRAP, REMAP, and IRAP+REMAP, Mantel’s test of matrix comparison with 1,000 permutations was performed. Similarity matrices of the IRAP+REMAP data were used to establish a Dcenter matrix and were subsequently used in the principal coordinate analysis (PCoA) to resolve patterns of genetic relationships among the 101 genotypes. All of these computations were implemented in NTsysPC 2.1 (Rohlf 2000). An analysis of molecular variance (AMOVA) using Genalex 6.4 (Peakall and Smouse 2006), to partition genetic variation within genotypes and between cultivars and lines, was based on the IRAP+REMAP data. The number of loci, percentage of polymorphic loci (PPL), number of alleles or loci with a frequency higher than or equal to 5%, number of private loci or alleles, number of less common loci with frequency lower than or equal to 25% and 50%, mean of expected heterozygosity (H e), number of effective alleles (N e), and Shannon’s information index (I) were calculated for each primer using Genalex 6.4 (Peakall and Smouse 2006). Those parameters were also measured for lines, cultivars, and cultivars+lines using IRAP, REMAP, and IRAP+REMAP data. The efficiency of native and nonnative RTN-derived markers in polymorphism detection was compared by calculating total loci, polymorphic loci, PPL, H e, N e, and I separately for native and nonnative primers for cultivars, lines, and genotypes.
Results
RTN Activity and Polymorphism in T. aestivum Genome and IRAP Analysis
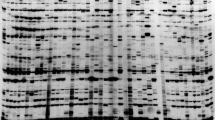
Of the tested IRAP primers, four single and five IRAP primer combinations (Table 1) generated scorable and polymorphic banding patterns among 101 bread wheat genotypes. Single primers Sukkula LTR1 (nonnative), Wilma-Bagy2, Daniela LTR2, and Fatima LTR produced polymorphic banding patterns. Most of the native IRAP primer combinations amplified no scorable and polymorphic banding patterns. Nine IRAP primers amplified 128 loci, of which 108 were polymorphic (84%). The frequency of all amplified loci was more than 5%. Primer Wilma-Bagy2 was 100% polymorphic (Fig. 1). Primer Daniela LTR2, a single native primer, showed the highest H e, N e, and I. The lowest values for those parameters, as well as for PPL, were obtained for the nonnative primer combination Bare1 LTR-Sukkula LTR2.

Polymorphism detected by a IRAP primer Wilma-Bagy2 and b REMAP primer combination Daniela LTR2-UBC815. a Cultivars in lane 1 Adl, 2 Dez, 3 Shahpasand, 4 Mahdavi, 5 Arya, 6 Arta, 7 Atrak, 8 Shahryar, 9 Niknejad, 10 Shiraz; breeding lines in lane 11 159, 12 183, 13 1717, 14 167, 15 175, 16 146, 17 140, 18 25, 19 137, 20 164. b Breeding lines in lane 1 140, 2 25, 3 137, 4 164, 5 169, 6 145, 7 149, 8 8, 9 21, 10 121, 11 27, 12 32, 13 38, 14 114, 15 125, 16 19, 17 110, 18 20, 19 28, 20 115. Lane M 1 kb O’GeneRuler DNA ladder (Fermentas) in base pairs
The Dice coefficients of genetic similarity using IRAP data averaged 0.81 for lines, 0.8 for cultivars, and 0.8 for lines+cultivars. The greatest genetic similarity was 0.98, between lines 12 and 177, and the least was 0.68, between Chenab and 608. A complete linkage dendrogram using 128 IRAP loci divided the individuals into five groups (Fig. 2). Lines were mostly in the vicinity of each other in the dendrogram, as was also observed for cultivars.



Complete linkage dendrograms of 101 bread wheat cultivars and breeding lines using a 128 IRAP loci, b 263 REMAP loci, and c 391 IRAP+REMAP loci. All dendrograms based on Dice similarity coefficient
REMAP Analysis
Of the 88 tested REMAP primer combinations, 20 generated 263 scorable loci, and 240 (91%) of the loci were polymorphic (Table 1). Sukkula LTR2 and Bare1 LTR, as nonnative primers, produced no polymorphic banding patterns in REMAP reactions. All native primer combinations produced polymorphic banding patterns in REMAP reactions. The average of REMAP polymorphic loci was 12 per primer. The size range of the amplified loci was 75–2,000 bp (Table 1). Polymorphism detected by the REMAP primer combination Daniela LTR2-UBC815 is presented in Fig. 1. The Dice genetic similarity coefficient based on REMAP loci ranged from 0.63 (between lines 159 and 608) to 0.92 (between lines 122 and 133), averaging 0.80. Five main clusters were identified based on REMAP data using a complete linkage algorithm (Fig. 2). Similar to the IRAP dendrogram, cultivars or lines were near each other in the same cluster.
Combined Data Analysis
The N e, I, H e, and PPL comparison among cultivars and breeding lines was based on IRAP, REMAP, and IRAP+REMAP analysis. In all three, the values calculated for breeding lines were more than those of cultivars, showing a high level of RTN insertional polymorphism and transpositional activity in the breeding lines (Table 2). The parameters were calculated separately for native and nonnative primers, to compare their efficiency in detecting polymorphism and diversity. No remarkable difference was detected between the two groups, suggesting insertional activity of the nonnative RTNs used in the bread wheat genome (Table 3).
Cophenetic matrices of IRAP and REMAP markers were significantly correlated with the IRAP+REMAP data, but correlations between IRAP and REMAP cophenetic matrices were not significant (r = 0.13). Hence, the data of IRAP+REMAP markers were used to build a dendrogram to evaluate the power of both techniques when combined. The Dice genetic similarity coefficient of the IRAP+REMAP data ranged from 0.68 (between lines 151 and 1661) to 0.92 (between lines 122 and 133), with a mean value of 0.80. Cluster analysis using the Dice similarity coefficient and a complete linkage algorithm revealed five groups (Fig. 2; Table 4). Similar to the results from IRAP and REMAP markers, the cultivars and breeding lines were in proximity to each other and in the same group or subgroup. Most of the cultivars were located in cluster II. Principal coordinate analysis confirmed the results obtained from cluster analysis, although PCoA1 and PCoA2 accounted for only 11% of the total variation in the genotypes studied (Fig. 3).

Two-dimensional plot of the genetic relationships among 101 bread wheat lines and cultivars as revealed by principal coordinate analysis using IRAP+REMAP data
Discussion
RTN Transpositional Activity and Polymorphism in Iranian Bread Wheat Genotypes
To our knowledge, this is the first report of IRAP and REMAP-based assessments of RTN transpositional activity in Iranian bread wheat cultivars and breeding lines. Nine IRAP and 20 REMAP primers were shown to amplify polymorphic and discernible banding patterns and were applied to study the RTN transpositional activity and the genetic diversity among 101 Iranian bread wheat cultivars and breeding lines. In our study, the high percentage of polymorphism detected by both IRAP (84%) and REMAP (91%) markers suggested the activities of the RTN families used in the Iranian bread wheat genome. Much polymorphism was detected by REMAPs, since the bands produced by REMAP are better than those produced by IRAP at showing the number of LTRs present in the genome (Kalendar et al. 1999), explaining the high average percentage of REMAP polymorphism. RTNs may integrate in principle in either orientation into the genome; hence, any two members of one family or different RTN families may be found head to head, tail to tail, or head to tail (Kalendar et al. 1999; Abdollahi Mandoulakani et al. 2012). Therefore, combined primers from LTR ends of different RTN families were used in IRAP reactions to increase the probability of finding bands. Single IRAP primers Wilma-Bagy2, Daniela LTR2, Fatima LTR, and Sukkula LTR1 produced polymorphic and distinguishable banding patterns, indicating the presence and transpositional activity of these elements in the bread wheat genome. The detection of a large number of IRAP and REMAP fragments generated by Wilma-Bagy2 primers revealed high copy numbers and a possible role of this RTN family in the construction, organization, and evolution of the bread wheat genome that may be of use in certain applications. Primers Bare1 LTR and 5LTR amplified no bands as single IRAP primers, but they generated bands in IRAP reactions as primer combinations with Sukkula and Daniela. This might reflect the insertion of Bare1 in or near these elements and the absence of local clustering of them in the wheat genome. The insertion of Bare1 near the Sukkula and other RTNs has been previously found in the genome of Portuguese bread wheat cultivars (Carvalho et al. 2010) and Aegilops tauschii (Saeidi et al. 2008). The applicability of the barley RTNs for genome analysis in the genera Aegilops, Hordeum, and Triticum has been previously reported (Kalendar et al. 1999; Queen et al. 2004). Vicient et al. (1999) stated that grasses share families of transcriptionally, translationally, and integrationally active RTNs. Our study showed that Bare1 and Sukkula, as nonnative RTNs, have relatives in the Iranian wheat genome and are transpositionally active, as demonstrated earlier (Carvalho et al. 2010; Saeidi et al. 2008). Wis2-1A has been previously considered an ortholog of Bare1 in the wheat genome (Vicient et al. 1999, 2005; Muniz et al. 2001), but primers based on both families produced different banding patterns in IRAP and REMAP reactions in our study, suggesting the divergence of these two families in the Iranian wheat genome. Therefore, Wis2-1A and Bare1 might be defined as separate RTN families. Several investigations have demonstrated that primers based on LTR sequences of RTN families can be readily used across species lines, among closely related genera, and even sometimes between plant families (Lou and Chen 2007; Abdollahi Mandoulakani et al. 2012). Wis2-1A LTR, Wis LTR1, and Wis LTR2, as native RTNs, did not produce bands singly or in combination with other RTN families in IRAP reactions, but they amplified bands in REMAP reactions, probably demonstrating their presence in the wheat genome as solo LTRs and the preferential insertion of this RTN family near SSR motifs in the wheat genome. Most of the RTNs used here produced bands in REMAP reactions, showing their insertion near or in SSR motifs, except for Bare1. This might show preferential insertion of Bare1 in SSR motifs in the wheat genome. Insertions of RTN families in the vicinity of microsatellites have been reported previously in barley, wheat, and alfalfa (Vicient et al. 1999, 2005; Carvalho et al. 2010; Abdollahi Mandoulakani et al. 2012). The polymorphism and diversity detected by native RTN-based primers used in IRAP and REMAP reactions in the genotypes studied here were not considerably different from those of nonnatives (Table 3), demonstrating the presence and insertional activity of the nonnative RTNs used in the wheat genome and the likely close phylogenetic relationship of wheat and barley. Abdollahi Mandoulakani et al. (2012) reported that IRAP and REMAP primers based on RTNs isolated from Medicago sativa show more polymorphism and are more informative than those based on nonnative RTNs (LORE1 and LORE2 isolated from Lotus japonicus). The difference in the number of native and nonnative primers used in the two investigations might be responsible for the contrasting results.
Genetic Relationship Among Studied Genotypes
The correlations estimated among the three cophenetic matrices generated from the IRAP, REMAP, and IRAP+REMAP dendrograms evidenced a relatively high and significant correlation of IRAP and REMAP with IRAP+REMAP. The matrices estimated by the techniques individually, however, revealed low and nonsignificant correlations (r = 0.13). These results suggest that DNA regions amplified by REMAP primers are different from those covered by IRAP, similar to the findings in barley (Kalendar et al. 1999), rice (Branco et al. 2007), and alfalfa (Abdollahi Mandoulakani et al. 2012). Hence, combined analysis of IRAP+REMAP markers was applied to reveal the association among cultivars and lines and identification of heterotic groups among 101 bread wheat genotypes. The low level of genetic variation detected among these genotypes (I = 0.5, H e = 0.34, and Dice similarity coefficient = 0.8) might be ascribed to the self-pollinating nature of wheat. Also, various wheat breeding programs in recent decades have reduced the genetic variability of Iranian commercial wheat cultivars. Evidence for this can be seen in the epidemic of wheat yellow rust (Puccinia striiformis f. sp. tritici) in most regions of Iran in the three last years. Mohammadi et al. (2009) reported high values of SSR-based gene diversity for 27 Iranian local commercial (PIC = 0.7) and adapted (0.66) wheat cultivars. The differences between those results and our study may be due to differences in the numbers of samples and the marker systems used. However, complete linkage-cluster analysis based on the Dice similarity coefficient using 391 IRAP+REMAP loci identified five heterotic groups (Table 4). Cultivars were distributed in three groups (1, 2, and 3) and breeding lines in four groups (1, 2, 4, and 5). Most cultivars as well as breeding lines were located in group 2. The remaining cultivars were in groups 1 and 3. Clusters 4 and 5 consisted of breeding lines only. All commercial cultivars were in the vicinity of each other in the same subgroup, as was observed for breeding lines. The largest genetic distance was found between lines 151 and 1661. These two lines or lines and cultivars in different heterotic groups could be introduced as potentially suitable crossing parents in wheat breeding programs to explore heterosis as well as to produce mapping populations, provided that factors such as combining ability and other agronomically important traits are taken into account. Now, morphological and physiological traits of these genotypes are recorded in two regions for two years to identify reliable molecular markers associated with agronomically and economically important traits.
In conclusion, the application of RTN-based markers can be a valuable tool for wheat breeders, as it was for barley (Kalendar et al. 1999), citrus (Biswas et al. 2010), and alfalfa (Abdollahi Mandoulakani et al. 2012). In comparison with other DNA markers, they might be even more informative and reliable for wheat researchers, as 40–60% of the wheat genome comprises different RTN families (Muniz et al. 2001). In wheat, where RTN LTRs have been identified, the use of a multi-RTN approach in the molecular analysis is particularly informative, because each RTN may have a unique transpositional history (Kumar and Bennetzen 1999), and thus wider genome coverage is achieved. Similarly, the density of markers on a recombinational map can be increased by pooling data from several RTNs. Using different family elements offers the possibility of increasing phylogenetic resolution on the basis of such differences. Also, a combination of ancient and active retrotransposons can provide a comprehensive evaluation of the evolutionary history of a genome and the relationships between taxa. In the current study, IRAP and REMAP markers have demonstrated great advantages, with feasible operation, a simple technique, and high reliability for the study of genetic diversity and relationships among wheat cultivars and breeding lines. They were sufficiently polymorphic to allow detection of intervarietal polymorphism and heterotic groups in the wheat germplasm used.
References
Abdollahi Mandoulakani B, Piri Y, Darvishzadeh R, Bernoosi I, Jafari M (2012) Retroelement insertional polymorphism and genetic diversity in Medicago sativa populations revealed by IRAP and REMAP markers. Plant Mol Biol Rep 30:286–296
Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, Albright LM, Coen DM, Varki A (1995) Current protocols in molecular biology. Jon Wiley, New York
Biswas MK, Baig MNR, Cheng YJ, Deng XX (2010) Retrotransposon based genetic similarity within the genus citrus and its relatives. Genet Resour Crop Evol 7:963–972
Branco JSC, Vieira EA, Malone G, Kopp MM, Malone E, Bernardes A, Mistura CC, Carvalho FIF, Oliveira CA (2007) IRAP and REMAP assessments of genetic similarity in rice. J Appl Genet 48:107–113
Carvalho A, Guedes-Pinto H, Martins-Lopes P, Lima-Brito J (2010) Genetic variability of old Portuguese bread wheat cultivars assayed by IRAP and REMAP markers. Ann Appl Biol 3:337–345
Ellis THN, Poyser SJ, Knox MR, Vershinin AV, Ambrose MJ (1998) Ty1-copia class retrotransposon insertion site polymorphism for linkage and diversity analysis in pea. Mol Gen Genet 260:9–19
Fernie AR, Tadmor Y, Zamir D (2006) Natural genetic variation for improving crop quality. Curr Opin Plant Biol 9:196–202
Kalendar R, Grob T, Regina M, Suoniemi A, Schulman A (1999) IRAP and REMAP: two retrotransposon-based DNA fingerprinting techniques. Theor Appl Genet 98:704–711
Kalendar R, Flavell AJ, Ellis THN, Sjakste T, Moisy C, Schulman AH (2011) Analysis of plant diversity with retrotransposon-based molecular markers. Heredity 106:520–530
Kumar A, Bennetzen JL (1999) Plant retrotransposons. Annu Rev Genet 33:479–532
Lou Q, Chen J (2007) Ty1-copia retrotransposon-based SSAP marker development and its potential in the genetic study of cucurbits. Genome 50:802–810
Mohammadi SA, Khodarahmi M, Jamalirad S, Jalal Kamali MR (2009) Genetic diversity in a collection of old and new bread wheat cultivars from Iran as revealed by simple sequence repeat-based analysis. Ann Appl Biol 154:67–76
Muniz L, Cuadrado A, Jouve N, Gonzalez JM (2001) The detection, cloning, and characterisation of WIS2-1A retrotransposon-like sequences in Triticum aestivum L. and × Triticosecale Wittmack and an examination of their evolution in related Triticeae. Genome 44:979–989
Peakall R, Smouse PE (2006) Genalex 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Queen RA, Gribbon BM, James C, Jack P, Falvell AJ (2004) Retrotransposon-based molecular markers for linkage and genetic diversity analysis in wheat. Mol Genet Genomics 271:91–97
Rohlf FJ (2000) NTSYS-pc: numerical taxonomy and multivariate analysis system, version 2.1. Exeter Software, New York
Roussel V, Koenig J, Beckert M, Balfourier F (2004) Molecular diversity in French bread wheat accessions related to temporal trends and breeding programs. Theor Appl Genet 108:920–930
Roussel V, Leisova L, Exbrayat F, Stehno Z, Balfourier F (2005) SSR allelic diversity changes in 480 European bread wheat varieties released from 1840 to 2000. Theor Appl Genet 111:162–170
Saeidi H, Rahiminejad MR, Heslop-Harrison JS (2008) Retroelement insertional polymorphisms, diversity and phylogeography within diploid, D-genome Aegilops tauschii (Triticeae, Poaceae) sub-taxa in Iran. Ann Bot 101:855–861
Salamini F, Ozkan H, Brandolini A, Schafer-Pregl R, Martin W (2002) Genetics and geography of wild cereal domestication in the near east. Nat Rev Genet 3:429–441
San Miguel P, Tikhonov A, Jin Y-K, Motchoulskaia N, Zakharov D, Melake-Berhan A, Springer PS, Edwards KJ, Lee M, Avramova Z, Bennetzen JL (1996) Nested retrotransposons in the intergenic regions of the maize genome. Science 274:765–768
Smykal P (2006) Development of an efficient retrotransposon-based fingerprinting method for rapid pea variety identification. J Appl Genet 47:221–230
Tam SM, Mhiri C, Vogelaar A, Kerkveld M, Pearce SR, Le Grandbastien MA (2005) Comparative analyses of genetic diversities within tomato and pepper collections detected by retrotransposon-based SSAP, AFLP and SSR. Theor Appl Genet 110:819–831
Tanksley SD, McCouch SR (1997) Seed banks and molecular maps: unlocking genetic potential from the wild. Science 277:1063–1066
Vicient CM, Kalendar R, Anamthawat-Jonnson K, Suoniemi A, Schulman AH (1999) Structure, functionality, and evolution of the BARE-1 retrotransposon of barley. Genetica 107:53–63
Vicient CM, Kalendar R, Schulman AH (2005) Variability, recombination and mosaic evolution of the barley BARE-1 retrotransposon. J Mol Evol 61:275–291
Acknowledgments
The authors are grateful to the Faculty of Agriculture and the Institute of Biotechnology, Urmia University, Iran, for financial support of the work. The Seed and Plant Improvement Institute is thanked for providing seed materials. Dr. R. Kalendar is thanked for his helpful discussions on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Nasri, S., Abdollahi Mandoulakani, B., Darvishzadeh, R. et al. Retrotransposon Insertional Polymorphism in Iranian Bread Wheat Cultivars and Breeding Lines Revealed by IRAP and REMAP Markers. Biochem Genet 51, 927–943 (2013). https://doi.org/10.1007/s10528-013-9618-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10528-013-9618-5