
Abstract
Medical advancements have extended human life expectancy, which is not always accompanied by an improved quality of life or healthspan. A decline in muscle mass and function is a consequence of ageing and can result in a loss of independence in elderly individuals while increasing their risk of falls. Multiple cellular pathways have been implicated in age-related muscle atrophy, including the contribution of reactive oxygen species (ROS) and disrupted redox signalling. Aberrant levels of ROS disrupts the redox environment in older muscle, potentially disrupting cellular signalling and in some cases blunting the adaptive response to exercise. Age-related muscle atrophy is associated with disrupted mitochondrial content and function, one of the hallmarks of age-related diseases. There is a critical link between abnormal ROS generation and dysfunctional mitochondrial dynamics including mitochondrial biogenesis, fusion and fission. In order to develop effective treatments or preventative strategies, it is important to gain a comprehensive understanding of the mechanistic pathways implicated in age associated loss of muscle.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
An extended life span does not automatically imply preservation of the quality of life in later years and is associated with increasing incidences of age-related diseases such as osteoporosis, neurodegenerative disorders and diabetes (Partridge et al. 2018). The age related loss of muscle mass and function or sarcopenia can result in a 30–50% decrease in skeletal muscle mass and accompanied by a reduction in muscular force in the elderly population (Marzetti et al. 2013). Reductions in skeletal mass and function can result in a loss of independence, a reduced quality of life, increased hospital visits and a predisposition to injuries and falls (Valenzuela et al. 2019). Inactivity including bedrest as a result of illness can further exacerbate muscle atrophy and augments the vulnerability of an individual to further injuries and potential infections. In order to develop effective treatments or preventative strategies with the ultimate aim to reduce frailty in the elderly and maintain muscular function, it is important to understand the mechanistic pathways implicated in the age associated loss of muscle (Marzetti et al. 2013).
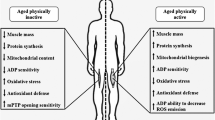
Research to date suggests the implication of multiple cellular pathways in age-related muscle atrophy. Particular emphasis has been placed on the involvement of reactive oxygen species (ROS) and a disrupted redox environment in muscle from older individuals with subsequent effects on cellular signalling and the adaptive response to exercise (Jackson 2016; McDonagh et al. 2014). ROS include, the hydroxyl radical (OH·), superoxide (O2·−), nitric oxide (NO), hydrogen peroxide (H2O2) and peroxynitrite (ONOO−), all of which can be generated endogenously either directly or indirectly as a consequence of metabolic processes within the cell (Reczek and Chandel 2015). Endogenous generation of ROS in skeletal muscle following contractile activity was first demonstrated using electron spin resonance (Davies et al. 1982; Jackson et al. 1985). An ex vivo study using electrically stimulated flexor digitorum brevis (FDB) fibres has demonstrated that the NADPH oxidase isoform 2 (NOX2) located on the sarcolemma or transverse t-tubules, is primarily responsible for contraction induced ROS generation (Sakellariou et al. 2013). The contractile induced NOX2 dependent increase in ROS has recently been demonstrated to be required for GLUT4 translocation in muscle fibres (Henríquez-Olguin et al. 2019). ROS are key signalling molecules that are specifically generated during muscle contractions and are required for the correct adaptive response to exercise (for review see (Goljanek-Whysall et al. 2016; Henriquez-Olguin et al. 2020; Margaritelis et al. 2020). The adaptive response to skeletal muscle contractions ensures that endogenous ROS generation is maintained within a homeostatic range for signalling, an acute increase in response to contractions as opposed to chronically elevated levels as a result of ageing. An imbalance in ROS concentrations, disturbs the homeostatic balance resulting in maladaptation and pathophysiological states (Alleman et al. 2014) (Fig. 1). The elevated basal level of ROS in muscle from older sedentary subjects could result in a disruption of the signalling pathways during muscle contractions. Consequently there was a blunted response reported with age to stimulate the signalling pathways required for the adaptations of skeletal muscle to exercise observed in younger individuals, senior sportsmen were able to adapt to some but not all of the parameters tested (Cobley et al. 2014) This may be as a result of an altered redox environment but also the consequence of an accumulation of damage, ultimately resulting in a narrowing of the homeostatic range as we age. As a result, an oxidative or reductive stress that may have resulted in a beneficial adaptive response in younger individuals could result in a damaging effect as we age (Fig. 1).

An individual can experience pulsatile bouts of oxidative (or reductive) stress in skeletal muscles particularly in response to bouts of exercise. Intermittent periods of ROS-induced stress within homeostatic range induces beneficial adaptive responses in skeletal muscle myofibres, associated with improved antioxidant capacity and cellular quality control adaptations. In sedentary older individuals there is a narrowing of the homeostatic range. Previously beneficial elevated levels of ROS can push the myofibre outside the homeostatic range, inducting deleterious consequences and overall, a failure to adapt to bouts of oxidative stress
Redox signalling and skeletal muscle
The oxidative stress theory of aging has been refined and there has been a greater appreciation of both the beneficial and deleterious effects of endogenous ROS generation in cell physiology (Jones 2015). The beneficial roles of ROS in redox signalling has been supported by studies using supplementation with Vitamin C and E, which can dampen some of the beneficial adaptive responses to exercise (Gomez-Cabrera et al. 2008, 2012; Paulsen et al. 2014; Ristow et al. 2009). Disrupted redox signalling can result in the inhibition of anabolic signalling through the mechanistic target of rapamycin complex 1 (mTORC1) after resistance exercise, that has been reported in a number of human studies after ingestion of Vitamin C and E (Bjørnsen et al. 2016; Paulsen et al. 2014).
It has also been demonstrated that repair of the myocyte plasma membrane post injury was exclusively dependent on increased ROS production as a result of increased mitochondrial Ca2+ via the mitochondrial Ca2+ uniporter (MCU) (Horn et al. 2017). Following injury, Ca2+ enters mitochondria via the MCU stimulating the production of ROS which then activates guanosine triphosphatase RhoA (GTPase RhoA), leading to the accumulation of F-actin at the location of the injury subsequently initiating repair of the plasma membrane (PM) (Horn et al. 2017). Inhibition of ROS generation triggered by MCU Ca2+ uptake resulted in compromised repair of the PM accompanied by a greater extent of skeletal myofibre damage and deterioration of the force produced. This study highlights the need for Ca2+ induced mitochondrial ROS production to ensure the repair of the myofibril PM (Horn et al. 2017). Furthermore, a muscle specific MCU1 knockout mouse model resulted in reduced mitochondrial Ca2+ uptake during contractile activity, impaired repair of the plasma membrane and ultimately muscle atrophy (Debattisti et al. 2019). The site specific nature of endogenous ROS generation coupled with the resources required for their generation would indicate that exercise induced ROS signalling has an important biological role (Margaritelis et al. 2016).
Adaptive response to exercise
The elevated production of ROS during contractile activity upregulates the activity of transcription factors, nuclear factor-κB (NF-κB), activator protein 1 (AP-1) and nuclear factor erythroid 2-related factor 2 (Nrf2), to augment the activity of antioxidant enzymes, leading to exercise induced adaptations that protect the muscle from damage during periods of oxidative stress (Done et al. 2016; Vasilaki et al. 2006; Yamada et al. 2019). This upregulation of transcriptional activity appears to be the basis for the cellular based adaptive protective mechanism against the potential harmful effects of oxidative stress or elevated ROS concentrations. Skeletal muscle from old mice post contractions and in older men after exercise have attenuated activation of antioxidant transcription factors required for induction of enzymes involved in redox homeostasis (Done et al. 2016; Done and Traustadottir 2016; Vasilaki et al. 2006), It would suggest that due to an altered redox environment in aging muscle, there may be chronic activation of pro-inflammatory transcription factors such as NF-κB and attenuation of anti-oxidant transcription factors such as Nrf2 resulting in the blunted response of muscle to exercise in old mice. Under basal condition NF-κB is sequestered in the cytoplasm by IκB kinases, a phosphorylation signalling cascade results in degradation of the regulatory proteins allowing translocation of NF-κB to the nucleus and transcription of inflammation and stress response genes. ROS can both activate and inhibit NF-κB signalling (for review see (Morgan and Liu 2011)) as many of the canonical regulatory proteins are redox sensitive e.g. Cys50 of p50 is sensitive to oxidation (Matthews et al. 1992). There is also considerable crosstalk between NF-kB and exercise induced ROS activation of C-Jun N-terminal kinase (JNK) and p38-MAPK kinase signalling, for instance exercise induced ROS can stimulate kinase signalling and glucose uptake in muscle (Chambers et al. 2009; Henríquez-Olguin et al. 2019). Whether these regulatory enzymes are directly oxidised in response to an elevation of ROS concentrations or via intermediary proteins such as the Peroxiredoxins in a redox relay mechanism has yet to be determined in skeletal muscle. Many of these studies highlight the delicate balance between endogenous ROS generation and redox signalling in skeletal muscle ageing and exercise, a controlled increase in ROS during exercise for adaptation and the extensive damage triggered by a chronic, global elevation of cellular ROS production with age.
Mitochondria, ROS and exercise
Exercise induced activation of redox sensitive transcription factors and cytoprotective proteins is also accompanied by mitochondrial adaptations with increased respiratory capacity and mitochondrial protein content (Holloszy 1967; Mansueto et al. 2017). The increase in mitochondrial content is largely dependent on peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), which is sensitive to changes in the redox environment (Puigserver and Spiegelman 2003). ROS concentrations and mitochondrial function are intrinsically linked and electron leakage from mitochondria has been considered a major source of non-contractile generated ROS (Muller et al. 2007). In skeletal muscle under quiescent or resting conditions associated with low ATP demand coupled with increased [NADH], there are increases in mitochondrial O2·− production primarily from mitochondrial complexes I and III (Goncalves et al. 2015). However, using mitochondria isolated from rat skeletal muscle that were incubated using substrates to mimic exercise, ROS generation was lower (Goncalves et al. 2015). The presence of dysfunctional mitochondria particularly in ageing, can result in mitochondrial swelling, loss of cristae, destruction of the inner membrane and impaired respiration (Bratic and Larsson 2013). However, recently using the sensitive H2O2 probe HyPer7, Belousov and colleagues demonstrated that H2O2 can pass from the cytosol to mitochondria but not from mitochondria to the cytosol, only in dysfunctional mitochondria or where the mitochondrial reducing power has been eliminated results in elevated levels of cytosolic ROS (Pak et al. 2020). Mitochondria are positioned at a critical metabolic junction coordinating cellular signalling essential for oxidative phosphorylation, amino acid biosynthesis, Fe–S cluster biogenesis but can also initiate cell death via apoptosis. Elevated levels of ROS may disrupt signalling pathways that contribute to disrupted mitochondrial biogenesis and remodelling, increasing the tendency of the cells to undergo apoptosis and hence contribute to atrophy of aging muscle. In post mitotic tissues such as skeletal muscle both mitochondrial function and dynamics have a central role in the pathogenesis of age-related skeletal muscle atrophy and loss of function (Calvani et al. 2013). The promotion or disruption of mitochondrial dynamics during exercise and aging has also been associated with changes in expression at both the transcriptional and post-transcriptional levels (Ljubicic et al. 2010; Zinovkina and Zinovkin 2015).
Mitochondrial dysfunction in aging
Dysfunctional mitochondria are one of the main causative agents contributing to the aging phenotype (Lopez-Otin et al. 2013). The loss of mitochondrial content in skeletal muscle with age is well documented in a variety of different muscle fibre types (Holloszy et al. 1991; St-Jean-Pelletier et al. 2017). Mitochondrial dysfunction is classified as any irregularities in normal mitochondrial processes including regulation of Ca2+ concentrations, ATP generation, synthesis of lipids and ROS detoxification (Brand and Nicholls 2011). The decline in ATP production is accompanied by elevated ROS release from a defective electron transport chain impacting on the efficiency of the organelle (Chabi et al. 2008). The production of ROS via electron leakage is dependent on the assembly of super-complexes (Goncalves et al. 2015). The subsequent degradation of these super-complexes as we get older is thought to be responsible for non-reversible and excessive concentrations of ROS associated with the ageing phenotype (Genova and Lenaz 2015).
As we age skeletal muscle bioenergetics are not as efficient in maintaining the same respiratory function as younger individuals and are more susceptible to mitochondrial uncoupling (Gouspillou et al. 2014). This inefficiency is thought to be brought about by (1) a reduction in the biogenesis of new mitochondria combined with (2) the accumulation of defective mitochondria in the muscle tissues due to a disequilibrium between mitochondrial fusion and fission (Hepple 2014). Most mitochondrial proteins that are synthesised in the cytoplasm are imported into mitochondria through dedicated protein channels such as TOM20 that recognises a mitochondrial targeting sequence (Abe et al. 2000). Mitochondria possess their own genome which encodes 37 genes, 2 for ribosomal RNAs, 22 for transfer RNAs and 13 which code for necessary proteins required for the electron transport chain. Of these 13 polypeptides, 1 is a subunit for complex III, 2 subunits of ATP synthase, 3 subunits of complex IV and 7 subunits of complex I (for review see Hepple 2014). Despite the fact that mitochondrial DNA only codes for a limited number of the proteins contained in the mitochondria, they are essential for a functional electron transport chain. As we age mitochondrial DNA has been reported to acquire harmful mutations which in turn impairs the synthesis of new mitochondria and could be in part responsible for the reduction of mitochondrial biogenesis documented in aging muscle (Hiona et al. 2010). In addition, mitochondria are constantly being replaced and degraded which ultimately controls the quality of the organelle present in the tissue. This process is disrupted with age and related to the accumulation of mitochondria with atypical function associated with aging (Hood et al. 2019). There are a multitude of signalling pathways which precisely regulate the processes of mitochondrial biogenesis, fusion, fission and mitophagy. In skeletal muscle aging, many of these systems become dysregulated, resulting in aggregations of inefficient and damaged mitochondria which are partially responsible for the debilitating effects associated with aging (for review see Hood et al. 2019).
Mitochondrial biogenesis
In order to maintain a healthy mitochondrial population it is essential that damaged mitochondria are recognised, removed and appropriate mitochondrial biogenesis can occur (Hepple 2014). The complex task of mitochondrial biogenesis encompasses the formation and organisation of mitochondrial DNA, lipids and proteins, with these actions carefully co-ordinated by intricate interplay between mitochondrial and nuclear genomes. Although the coordination of the mitochondrial and nuclear genomes is carefully synchronized, mitochondrial DNA is independent of the nuclear genome. The task of ensuring a correct balance between the transcription of proteins in nuclear and mitochondrial genomes relies on PGC-1α, a transcriptional co-activator responsible for mitochondrial gene transcription (Vega et al. 2000; Wu et al. 1999). Exercise is a process with high oxidative demands and therefore a potent stimulus for additional mitochondrial biosynthesis (for review see Hood et al. 2019). Hence a good indication of PGC-1α activity is the ability of muscle to increase the biogenesis of mitochondria in response to exercise. The importance of PGC-1α was demonstrated when deletion of the PGC-1α gene in skeletal muscle of mice, resulted in a reduction in mitochondrial biogenesis markers in combination with a decline in overall performance (Handschin et al. 2007). The adaptive response of enhanced mitochondrial biogenesis following exercise was also dampened in PGC-1α knock out mice (Geng et al. 2010). Overexpression of PGC-1α enhanced the oxidative capacity and the presence of mitochondrial proteins especially in slow twitch oxidative muscle fibres (Lin et al. 2002). Collectively, this highlights the importance of PGC-1α in the transcription of mitochondrial proteins as a prerequisite for the formation of new mitochondria, especially in situations with increased demand for mitochondrial biosynthesis for example in response to exercise (Drake et al. 2016).
Following repeated bouts of exercise, the need for the formation of mitochondria is amplified, PGC-1α is activated and together with nuclear respiratory factor 1 (NRF1) and estrogen-related receptor α (ERRα), elevate the transcription rate of the majority of mitochondrial genes (Safdar et al. 2011). The nuclear receptor interacting motif (LLKYL) facilitates the interaction of PGC-1α with ERRα initiating the transcription of mitochondrial genes encoded by the nuclear genome (Ichida et al. 2002).
PGC-1α and NRF1 interactions lead to a rise in the transcription of factor A, mitochondrial (TFAM), a mitochondrial transcription factor encoded by the nuclear genome and responsible for mitochondrial genome replication (Safdar et al. 2011; Wu et al. 1999). Exercise promotes the trafficking of PGC-1α to the mitochondria where it interacts with TFAM forming a network on the mitochondrial DNA at the location of the D loop constituent (Wu et al. 1999) (Fig. 2). This action stimulates mitochondrial biogenesis as it encourages the replication of mitochondrial DNA (Wu et al. 1999). The interaction between PGC-1α, NRF1 and ERRα, facilitate the transcriptional needs for successful biogenesis of mitochondria as seen in exercise, demonstrating effective coordination between two independent genomes (for review see Drake et al. 2016). The reduced capability to produce new mitochondria parallels the ageing process (Betik et al. 2009), if PCG-1α is not upregulated there is a subsequent lack of interactions with NRF1 or ERRα (Betik et al. 2009). Precursor proteins that under normal conditions are trafficked to the mitochondria to aid mitochondrial biogenesis undergo increased degradation which in turn impedes the upregulation of mitochondrial biogenesis (Huang et al. 2010).

Schematic illustration highlighting the various alterations with age in the regulation of mitochondrial dynamics including ROS generation, mitochondrial biosynthesis, fusion, fission and mitophagy in response to exercise
ROS is becoming increasingly important in our understanding of mitochondrial dysfunction in aging, with particular emphasis on regulating mitochondrial dynamics and Ca2+ handling by the MCU (Horn et al. 2017). The importance of ROS has been demonstrated in studies where an increase in ROS production was followed by an upsurge in NRF1 and TFAM expression in endothelial cells (Perez-de-Arce et al. 2005). This highlights the signalling role of ROS in forming a link between endogenous generation during contractile activity and the stimulation of mitochondrial biogenesis in skeletal muscle. There is evidence that indicates expression or activities of TFAM, PCG-1α and NRF1 are sensitive to alterations in the redox environment that can ultimately affect mitochondrial biogenesis (Radak et al. 2013).
Mitochondrial fusion and fission
Mitochondria are constantly undergoing turnover via fusion and fission, processes that are essential for the maintenance of a healthy population in skeletal muscle (Hood et al. 2019). Fusion of mitochondria involves joining individual mitochondria together via their mitochondrial membranes which in turn forms a new mitochondrial complex. Fission entails the separation of mitochondria resulting in the production of distinct mitochondrial units which are targeted for selected degradation (Palikaras et al. 2018).
Mitochondrial fusion occurs when two distinct locations on the reticulum of mitochondria fuse both their outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM), combining the content and membranes of the mitochondria involved (Karbowski et al. 2004). Similar to the role of PGC-1α in mitochondrial biogenesis, mitochondrial fusion relies on optic atrophy 1 (OPA1) and the transmembrane GTPases, mitofusion 1 and 2 (MFN1 and MFN2) (Fig. 2). The joining of the OMM is coordinated by the transmembrane proteins MFN1 and MFN2, and mitochondrial fusion is impaired with deletion of MFN1 and MFN2 (Song et al. 2009). It has been demonstrated that the interaction between PGC-1α and ERRα stimulates fusion by promoting the transcription of MFN1 and MFN2, and that the expression of all these regulatory genes are increased after exercise, although peaking at different time points (Cartoni et al. 2005). The importance of dynamin-related GTPase, OPA1, in mitochondrial fusion has been demonstrated when Opa1 is deleted and the fusion of the IMM in vitro is prevented (Song et al. 2009). Deletion of OPA1 is a lethal phenotype in mice induced by increased circulation of Fibroblast growth factor 21 (Fgf21), mitochondrial dysfunction and loss of myogenic stem cells (Tezze et al. 2017). Acute muscle specific deletion of Opa1 in mice results in disruption of mitochondrial morphology, ER stress and muscle atrophy (Tezze et al. 2017). There is a decrease in the expression of MFN1, MFN2 and OPA1 in skeletal muscle from old sedentary subjects, however their expression is maintained in senior sportsmen (Tezze et al. 2017).
Mitochondrial dynamics encompasses not only fusion but also mitochondrial fission for quality control of mitochondrial turnover. Damage to the membrane potential of mitochondria can be responsible for the initiation of fission, separating the mitochondrial reticulum from the injured mitochondria (Youle and van der Bliek 2012). Fission, in particular OMM fission, is controlled by GTPase dynamin-1-like protein 1 (DRP1), which is recruited to injured mitochondria upon mitochondrial membrane depolarisation (Smirnova et al. 1998). The mitochondrial fission 1 protein (FIS1) together with FIS1 adapter mitochondrial division 1 protein (MDV1) are responsible for the trafficking of DRP1 in GTPase DRP1 dependent mitochondrial fission (James et al. 2003). FIS1 is attached to the OMM, where a 6-helical bundle with tetratricopeptide repeat motifs is formed which facilitates interactions between DRP1 and MDV1 (Zhang et al. 2012) (Fig. 2). It has also been reported that the tail-anchored membrane protein mitochondrial fission factor 1 (MFF1) is involved in the trafficking of DRP1 when mitochondria become damaged, initiating fission and the removal of defective mitochondria (Otera et al. 2010). Once GTP is hydrolysed, mitochondrial fission is achieved when a helix is formed by DRP1 around the OMM, segregating the injured section of the mitochondria from the healthy mitochondria and targets it for degradation via mitophagy (Drake et al. 2016).
Similar to OPA1, constitutive deletion of DRP1 in mice is lethal while inducible muscle specific deletion results in mitochondrial dysfunction, muscle atrophy and degeneration (Favaro et al. 2019). The inducible muscle specific deletion of DRP1 resulted in the inhibition of mitophagy resulting in functionally abnormal large mitochondria, as well as activation of the unfolded protein response, increase in Ca2+ uptake and fibre atrophy (Favaro et al. 2019). Interestingly the same group generated a mouse inducible muscle specific double knockout for OPA1 and DRP1 (Romanello et al. 2019). Simultaneous deletion of DRP1 and OPA1 in the mouse model, alleviated the phenotype of OPA1 knockout mice, indicating that in the context of mitophagy inhibition of fission is dominant over inhibition of fusion (Romanello et al. 2019).
The removal of damaged mitochondria via mitophagy
Mitochondrial fission separates sections of the organelle which have become damaged to prevent the accumulation of defective mitochondria, hence acting as a quality control mechanism for mitochondria. The end fate of malfunctioning mitochondria undergoing fission is their targeted degradation by the lysosome, termed mitophagy. Mitophagy is a vital catabolic process that involves the recycling and salvaging of mitochondrial contents such as amino acids through the fusion of a lysosome with the phagosome, promoting the population of efficient mitochondria and preserving the quality of mitochondria remaining in the muscle (Yu and Long 2015). Mitophagy is successfully carried out when an autophagosome encapsulates the targeted mitochondria. There are a number of different pathways that can regulate mitophagy, including ubiquitin-mediated (Pink/Parkin pathway) and ubiquitin independent pathways (via mitophagy receptors on the outer mitochondrial membrane (e.g. BNIP3). However, the exact regulatory mechanisms of mitochondrial turnover remain to be fully understood (for reviews see Montava-Garriga and Ganley 2019 ; Palikaras et al. 2018).
The Pink/Parkin pathway of mitophagy has been well established. Following damaged induced loss of mitochondrial membrane potential, PINK1 is no longer imported into the mitochondrial matrix for degradation but accumulates on protein complexes on the OMM ,which are involved in protein transport out of the cytosol (Jin et al. 2010). As a result of PINK1 accumulation on the OMM, Parkin, an E3 ubiquitin ligase, is recruited to the injured mitochondria and interacts with the voltage-dependent anion channel (VDAC) and PINK1. Parkin ubiquitinates MFN1 and MFN2, inhibiting their function and stimulating the mitochondrial fission pathway (Chen and Dorn 2013; Narendra et al. 2008). Additionally, the ubiquitination of VDAC by Parkin, leads to the aggregation of MAP1LC3-interacting regions—(LIRs) containing p62/SQSTM1. The LIR attaches to MAP1LC3, signalling the removal of the tagged mitochondria via autophagosomes (Geisler et al. 2010). In skeletal muscle, the loss of Parkin results in decreased mitochondrial respiration, increased susceptibility of opening of the mitochondrial permeability pore and an increase in the fission protein Drp1 (Gouspillou et al. 2018).
Receptor mediated mitophagy involves LIR containing proteins such as BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3) and Nix, may prove essential to the recognition of dysfunctional mitochondria for degradation during exercise (for review see Palikaras et al. 2018). Mitophgy occurring via this mechanism involves attachment of the autophagosome to the OMM where BNIP3 and Nix have both accumulated allowing interaction with MAP1LC3. The importance of BNIP3 and Nix in mitophagy is observed when the LIR containing amino terminus of the Nix protein is deleted, resulting in a reduction in the removal of dysfunctional mitochondria (Palikaras et al. 2018). Current evidence highlights both BNIP3 and Nix involvement in the detection of damaged mitochondria for removal in skeletal muscle and could potentially be manipulated to address the accumulations of defective mitochondria often seen in aged muscle (Drake et al. 2016).
It has also been reported in skeletal muscle that in response to increased metabolic demand such as during exercise, mitophagy is regulated by 5’ AMP- activated protein kinase (AMPK) (Laker et al. 2017). AMPK interacts antagonistically with mTORC1 in order to promote mitophagy. The normal action of mTORC1 prevents the mitophagic process, firstly the presence of nutrients and Rag GTPases stimulates its translocation to the lysosome, inactivating the lysosome (Sancak et al. 2010), and secondly via the phosphorylation of the serine/threonine kinase unc51-like kinase (ULK1) along with the amalgamation of the ULK1, FIP200 and ATG13 network, impeding the activation of ULK1 (Sancak et al. 2010). AMPK acts to phosphorylate the mTORC1 component raptor and the upstream inhibitor tuberous sclerosis complex 2 tumour suppressor (TSC2) which impedes mTORC1 action, thus preventing trafficking of mTORC1 to the lysosome thereby encouraging mitophagy (Laplante and Sabatini 2012). Furthermore, AMPK also upregulates mitophagy by stimulating the activity of ULK1 (Fig. 2) (Laker et al. 2017), linking mitochondrial turnover with metabolic demand.
Disrupted mitochondrial dynamics in aging
An imbalance in mitochondrial fusion and fission has been reported to occur in skeletal muscle as we age (Hepple 2014). Evidence has indicated that levels of FIS1 along with activity of DRP1 declines in senescent cells (Mai et al. 2010), which may have implications in skeletal muscle during satellite cell activation and muscle regeneration. In models of muscle injury, such as the rotator cuff injury, with an associated loss of muscle force, there is a metabolic shift resulting in an accumulation of lipid and increased fibrosis associated with dysfunctional mitochondria and reduced fatty acid metabolism in myosteatosis (Gumucio et al. 2019). It is also interesting to note that in this study there was an accumulation of the authophagy adaptor protein p62, a classic indicator of autophagy impairment (Gumucio et al. 2019). Typically fragmentation of mitochondria, the loss in the mitochondrial membrane potential and elevated level of ROS generated, increases the rate of mitophagy (Schiavi and Ventura 2014). There is an aggregation of mitochondria sensitized to permeability transition in older muscular tissue (Gouspillou et al. 2014), suggesting that mitophagy is obstructed as we age because typically the loss of the membrane potential of mitochondria stimulates mitophagy (Twig et al. 2008). Parkin, which normally promotes fission and the removal of dysfunctional mitochondria via autophagosomes is also implicated in aging. Parkin knockout in mice results in reduced muscle force, decrease in mitochondrial respiration and increased susceptibility to the opening of the permeability transition pore (Gouspillou et al. 2018), while Parkin overexpression attenuates sarcopenia and results in muscle hypertrophy in adult skeletal muscle (Leduc-Gaudet et al. 2019). The coupling or integration between mitochondrial biogenesis and selective degradation via mitophagy is essential for the preservation of healthy skeletal muscle. Disruption of this balance can result in alterations in muscle bioenergetics and loss of muscle mass and function for example during ageing. In skeletal muscle from old mice, disruption of the mitophagic process can result in the accumulation of dysfunctional mitochondria, while improving the mitophagic flux results in a parallel increase in mitochondrial biogenesis improving overall mitochondrial function (Goljanek-Whysall et al. 2020). In C. elegans the age-related decline in mitophagy inhibits both the removal of dysfunctional mitochondria and impairs mitochondrial biogenesis (Palikaras et al. 2015). In our recent study we identified miR-181a as a potential regulator of mitochondrial dynamics and delivery of miR-181a to old mice improved mitochondrial content in muscle during ageing with physiologically-relevant consequences on myofibre size and force (Goljanek-Whysall et al. 2020). Although there are contrasting results on whether mitophagy flux is increased or decreased in skeletal muscle during ageing and exercise (Carter et al. 2018; Leduc-Gaudet et al. 2019; Romanello and Sandri 2015), it is clear that mitochondrial quality has an important impact on the health and bioenergetics of healthy skeletal muscle and is tightly regulated by mitochondrial biogenesis, fusion, fission and selective degradation of mitochondria. Current therapeutic options for mitochondrial diseases has focused on delivering mitochondrial components such as CoQ10, improving antioxidant capacity or increasing mitochondrial biogenesis by activating Nrf2 and PGC1α (for review see Zhang et al. 2020), pathways activated by redox dependent mechanisms during exercise.
Conclusions
Mitochondrial dysfunction and disrupted dynamics has a significant role to play in age related muscle atrophy and Sarcopenia. Consistent with the “Hallmarks of Aging” (Lopez-Otin et al. 2013), there is a significant loss of mitochondrial content in skeletal muscle with age that is accompanied by an increase in dysfunctional mitochondria (for review Hood et al. 2019). Moreover, accumulation of damaged or dysfunctional mitochondria increases the potential oxidation of contractile proteins in muscle and elevated ROS generation, altering mechanical function. There are a number of regulatory signalling pathways involved in mitochondrial dynamics including biogenesis, fusion and fission, one of the common factors that connect these pathways is redox signalling. In age-related muscle atrophy these regulatory pathways are disrupted, resulting in the accumulation of dysfunctional and damaged mitochondria, and accompanied by a reduction in the ability to stimulate mitochondrial biogenesis. There is an inability to further augment protective mechanisms and disrupted activation of transcription factors such as PGC-1α, Tfam, NF-κB and Nrf2, all thought to be brought about by chronic elevated levels of ROS in muscle from older sedentary subjects. Increased intracellular concentrations of ROS could potentially result in oxidative modifications of proteins, lipids and nucleic acids as compared to homeostatic concentrations for redox signalling. One of the outstanding issues in relation to redox signalling or the transfer of oxidising equivalents via ROS to selective targets, is the specificity of the signalling. The concept of localised endogenous ROS generation e.g. during contractile activity, diffusing through the cell to target a selective redox regulated enzyme or transcription factor has been questioned (Winterbourn 2015). One hypothesis that has been demonstrated in vitro is that redox signalling occurs via conserved intermediary proteins (Peroxiredoxins) that have evolved to specifically react with peroxides, transferring their oxidising equivalents to target proteins in a “redox relay” mechanism (Sobotta et al. 2015; Stocker et al. 2018). Similarly, apoptosis signalling factor-1 (ASK1), part of the p38 signalling pathway has been demonstrated to be indirectly oxidised by Peroxiredoxin 1 through the transfer of oxidising equivalents (Jarvis et al. 2012). A dedicated redox relay mechanism could potentially bring specificity to redox signalling, as ROS were originally considered as a non-specific oxidising agents. The altered redox environment in muscle fibres undergoing age-related atrophy could be a feedback response due to disrupted redox signalling as a result of aberrant mitochondrial ROS generation. The identification of the intermediary relay proteins and their targets would help in our understanding of the interplay between disrupted mitochondrial dynamics and redox signalling. In this review, we describe factors which may yield potential targets for therapeutic manipulation and in the case of sarcopenia, potentially ameliorate the debilitating loss of muscle mass and function as we age. A greater understanding of the factors and identification of intermediary proteins would allow a focus on developing strategies to combat the detrimental effects of aging such as the increased incidence of frailty, risk of falls and lack of independence.
References
Abe Y et al (2000) Structural basis of presequence recognition by the mitochondrial protein import receptor Tom20. Cell 100:551–560. https://doi.org/10.1016/s0092-8674(00)80691-1
Alleman RJ, Katunga LA, Nelson MA, Brown DA, Anderson EJ (2014) The “Goldilocks Zone” from a redox perspective-adaptive vs. deleterious responses to oxidative stress in striated. Muscle Front Physiol 5:358. https://doi.org/10.3389/fphys.2014.00358
Betik AC, Thomas MM, Wright KJ, Riel CD, Hepple RT (2009) Exercise training from late middle age until senescence does not attenuate the declines in skeletal muscle aerobic function. Am J Physiol Regul Integr Comp Physiol 297:R744–R755. https://doi.org/10.1152/ajpregu.90959.2008
Bjørnsen T et al (2016) Vitamin C and E supplementation blunts increases in total lean body mass in elderly men after strength training. Scand J Med Sci Sports 26:755–763. https://doi.org/10.1111/sms.12506
Brand MD, Nicholls DG (2011) Assessing mitochondrial dysfunction in cells. Biochem J 435:297–312. https://doi.org/10.1042/BJ20110162
Bratic A, Larsson NG (2013) The role of mitochondria in aging. J Clin Invest 123:951–957. https://doi.org/10.1172/JCI64125
Calvani R et al (2013) Mitochondrial pathways in sarcopenia of aging and disuse muscle atrophy. Biol Chem 394:393–414. https://doi.org/10.1515/hsz-2012-0247
Carter HN, Kim Y, Erlich AT, Zarrin-Khat D, Hood DA (2018) Autophagy and mitophagy flux in young and aged skeletal muscle following chronic contractile activity. J Physiol 596:3567–3584. https://doi.org/10.1113/JP275998
Cartoni R et al (2005) Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise. J Physiol 567:349–358. https://doi.org/10.1113/jphysiol.2005.092031
Chabi B, Ljubicic V, Menzies KJ, Huang JH, Saleem A, Hood DA (2008) Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 7:2–12. https://doi.org/10.1111/j.1474-9726.2007.00347.x
Chambers MA, Moylan JS, Smith JD, Goodyear LJ, Reid MB (2009) Stretch-stimulated glucose uptake in skeletal muscle is mediated by reactive oxygen species and p38 MAP-kinase. J Physiol 587:3363–3373. https://doi.org/10.1113/jphysiol.2008.165639
Chen Y, Dorn GW II (2013) PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340:471–475. https://doi.org/10.1126/science.1231031
Cobley JN et al (2014) Lifelong training preserves some redox-regulated adaptive responses after an acute exercise stimulus in aged human skeletal muscle. Free Radic Biol Med 70:23–32. https://doi.org/10.1016/j.freeradbiomed.2014.02.004
Davies KJ, Quintanilha AT, Brooks GA, Packer L (1982) Free radicals and tissue damage produced by exercise . Biochem Biophys Res Commun 107:1198–1205. https://doi.org/10.1016/s0006-291x(82)80124-1
Debattisti V et al (2019) Dysregulation of mitochondrial Ca(2+) uptake and sarcolemma repair underlie muscle weakness and wasting in patients and mice lacking MICU1. Cell Rep 29:1274-1286e.6. https://doi.org/10.1016/j.celrep.2019.09.063
Done AJ, Traustadottir T (2016) Nrf2 mediates redox adaptations to exercise. Redox Biol 10:191–199. https://doi.org/10.1016/j.redox.2016.10.003
Done AJ, Gage MJ, Nieto NC, Traustadottir T (2016) Exercise-induced Nrf2-signaling is impaired in aging. Free Radic Biol Med 96:130–138. https://doi.org/10.1016/j.freeradbiomed.2016.04.024
Drake JC, Wilson RJ, Yan Z (2016) Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. FASEB J 30(1):13–22. https://doi.org/10.1096/fj.15-276337
Favaro G et al (2019) DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat Commun 10:2576. https://doi.org/10.1038/s41467-019-10226-9
Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12:119–131. https://doi.org/10.1038/ncb2012
Geng T, Li P, Okutsu M, Yin X, Kwek J, Zhang M, Yan Z (2010) PGC-1alpha plays a functional role in exercise-induced mitochondrial biogenesis and angiogenesis but not fiber-type transformation in mouse skeletal muscle. Am J Physiol Cell Physiol 298:C572–C579. https://doi.org/10.1152/ajpcell.00481.2009
Genova ML, Lenaz G (2015) The interplay between respiratory supercomplexes and ROS in aging. Antioxid Redox Signal 23:208–238. https://doi.org/10.1089/ars.2014.6214
Goljanek-Whysall K, Iwanejko LA, Vasilaki A, Pekovic-Vaughan V, McDonagh B (2016) Ageing in relation to skeletal muscle dysfunction: redox homoeostasis to regulation of gene expression. Mamm Genome 27:341–357. https://doi.org/10.1007/s00335-016-9643-x
Goljanek-Whysall K, Soriano-Arroquia A, McCormick R, Chinda C, McDonagh B (2020) miR-181a regulates p62/SQSTM1, Parkin and protein DJ-1 promoting mitochondrial dynamics in skeletal muscle ageing. Aging Cell. https://doi.org/10.1111/acel.13140
Gomez-Cabrera MC et al (2008) Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am J Clin Nutr 87:142–149. https://doi.org/10.1093/ajcn/87.1.142
Gomez-Cabrera MC, Ristow M, Vina J (2012) Antioxidant supplements in exercise: worse than useless? Am J Physiol Endocrinol Metab 302:E476-477; author reply E478-479. https://doi.org/10.1152/ajpendo.00567.2011
Goncalves RL, Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Brand MD (2015) Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J Biol Chem 290:209–227. https://doi.org/10.1074/jbc.M114.619072
Gouspillou G et al (2014) Mitochondrial energetics is impaired in vivo in aged skeletal muscle. Aging Cell 13:39–48. https://doi.org/10.1111/acel.12147
Gouspillou G et al (2018) Protective role of Parkin in skeletal muscle contractile and mitochondrial function. J Physiol 596:2565–2579. https://doi.org/10.1113/JP275604
Gumucio JP, Qasawa AH, Ferrara PJ, Malik AN, Funai K, McDonagh B, Mendias CL (2019) Reduced mitochondrial lipid oxidation leads to fat accumulation in myosteatosis. FASEB J 33:7863–7881. https://doi.org/10.1096/fj.201802457RR
Handschin C et al (2007) Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J Biol Chem 282:30014–30021. https://doi.org/10.1074/jbc.M704817200
Henriquez-Olguin C, Meneses-Valdes R, Jensen TE (2020) Compartmentalized muscle redox signals controlling exercise metabolism - Current state, future challenges. Redox Biol. https://doi.org/10.1016/j.redox.2020.101473
Henríquez-Olguin C et al (2019) Cytosolic ROS production by NADPH oxidase 2 regulates muscle glucose uptake during exercise. Nat Commun 10:4623. https://doi.org/10.1038/s41467-019-12523-9
Hepple RT (2014) Mitochondrial involvement and impact in aging skeletal muscle. Front Aging Neurosci 6:211. https://doi.org/10.3389/fnagi.2014.00211
Hiona A et al (2010) Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS ONE 5:e11468. https://doi.org/10.1371/journal.pone.0011468
Holloszy JO (1967) Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem 242:2278–2282
Holloszy JO, Chen M, Cartee GD, Young JC (1991) Skeletal muscle atrophy in old rats: differential changes in the three fiber types. Mech Ageing Dev 60:199–213
Hood DA, Memme JM, Oliveira AN, Triolo M (2019) Maintenance of skeletal muscle mitochondria in health, exercise, and aging. Annu Rev Physiol 81:19–41. https://doi.org/10.1146/annurev-physiol-020518-114310
Horn A et al (2017) Mitochondrial redox signaling enables repair of injured skeletal muscle cells. Sci Signal 10:eaaj1978. https://doi.org/10.1126/scisignal.aaj1978
Huang JH, Joseph AM, Ljubicic V, Iqbal S, Hood DA (2010) Effect of age on the processing and import of matrix-destined mitochondrial proteins in skeletal muscle. J Gerontol A Biol Sci Med Sci 65:138–146. https://doi.org/10.1093/gerona/glp201
Ichida M, Nemoto S, Finkel T (2002) Identification of a specific molecular repressor of the peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1alpha). J Biol Chem 277:50991–50995. https://doi.org/10.1074/jbc.M210262200
Jackson MJ, Edwards RH, Symons MC (1985) Electron spin resonance studies of intact mammalian skeletal muscle. Biochim Biophys Acta 847:185–190
Jackson MJ (2016) Reactive oxygen species in sarcopenia: should we focus on excess oxidative damage or defective redox signalling? Mol Aspects Med 50:33–40. https://doi.org/10.1016/j.mam.2016.05.002
James DI, Parone PA, Mattenberger Y, Martinou JC (2003) hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem 278:36373–36379. https://doi.org/10.1074/jbc.M303758200
Jarvis RM, Hughes SM, Ledgerwood EC (2012) Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic Biol Med 53:1522–1530. https://doi.org/10.1016/j.freeradbiomed.2012.08.001
Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191:933–942. https://doi.org/10.1083/jcb.201008084
Jones DP (2015) Redox theory of aging. Redox Biol 5:71–79. https://doi.org/10.1016/j.redox.2015.03.004
Karbowski M, Arnoult D, Chen H, Chan DC, Smith CL, Youle RJ (2004) Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J Cell Biol 164:493–499. https://doi.org/10.1083/jcb.200309082
Laker RC et al (2017) Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun 8:548. https://doi.org/10.1038/s41467-017-00520-9
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149:274–293. https://doi.org/10.1016/j.cell.2012.03.017
Leduc-Gaudet JP, Reynaud O, Hussain SN, Gouspillou G (2019) Parkin overexpression protects from ageing-related loss of muscle mass and strength. J Physiol 597:1975–1991. https://doi.org/10.1113/JP277157
Lin J et al (2002) Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres . Nature 418:797–801. https://doi.org/10.1038/nature00904
Ljubicic V et al (2010) Transcriptional and post-transcriptional regulation of mitochondrial biogenesis in skeletal muscle: effects of exercise and aging. Biochim Biophys Acta 1800:223–234. https://doi.org/10.1016/j.bbagen.2009.07.031
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153:1194–1217. https://doi.org/10.1016/j.cell.2013.05.039
Mai S, Klinkenberg M, Auburger G, Bereiter-Hahn J, Jendrach M (2010) Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J Cell Sci 123:917–926. https://doi.org/10.1242/jcs.059246
Mansueto G et al (2017) Transcription factor EB controls metabolic flexibility during exercise. Cell Metab 25:182–196. https://doi.org/10.1016/j.cmet.2016.11.003
Margaritelis NV, Cobley JN, Paschalis V, Veskoukis AS, Theodorou AA, Kyparos A, Nikolaidis MG (2016) Principles for integrating reactive species into in vivo biological processes: examples from exercise physiology. Cell Signal 28:256–271. https://doi.org/10.1016/j.cellsig.2015.12.011
Margaritelis NV, Paschalis V, Theodorou AA, Kyparos A, Nikolaidis MG (2020) Redox basis of exercise physiology. Redox Biol. https://doi.org/10.1016/j.redox.2020.101499
Marzetti E, Calvani R, Cesari M, Buford TW, Lorenzi M, Behnke BJ, Leeuwenburgh C (2013) Mitochondrial dysfunction and sarcopenia of aging: from signaling pathways to clinical trials. Int J Biochem Cell Biol 45:2288–2301. https://doi.org/10.1016/j.biocel.2013.06.024
Matthews JR, Wakasugi N, Virelizier JL, Yodoi J, Hay RT (1992) Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res 20:3821–3830. https://doi.org/10.1093/nar/20.15.3821
McDonagh B, Sakellariou GK, Smith NT, Brownridge P, Jackson MJ (2014) Differential cysteine labeling and global label-free proteomics reveals an altered metabolic state in skeletal muscle aging. J Proteome Res 13:5008–5021. https://doi.org/10.1021/pr5006394
Montava-Garriga L, Ganley IG (2019) Outstanding questions in mitophagy: what we do and do not know. J Mol Biol. https://doi.org/10.1016/j.jmb.2019.06.032
Morgan MJ, Liu ZG (2011) Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res 21:103–115. https://doi.org/10.1038/cr.2010.178
Muller FL, Song W, Jang YC, Liu Y, Sabia M, Richardson A, Van Remmen H (2007) Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am J Physiol Regul Integr Comp Physiol 293:R1159–R1168. https://doi.org/10.1152/ajpregu.00767.2006
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803. https://doi.org/10.1083/jcb.200809125
Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K (2010) Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 191:1141–1158. https://doi.org/10.1083/jcb.201007152
Pak VV et al (2020) Ultrasensitive genetically encoded indicator for hydrogen peroxide identifies roles for the oxidant in cell migration and mitochondrial function. Cell Metab 31:642-653.e646. https://doi.org/10.1016/j.cmet.2020.02.003
Palikaras K, Lionaki E, Tavernarakis N (2015) Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521:525–528. https://doi.org/10.1038/nature14300
Palikaras K, Lionaki E, Tavernarakis N (2018) Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol 20:1013–1022. https://doi.org/10.1038/s41556-018-0176-2
Partridge L, Deelen J, Slagboom PE (2018) Facing up to the global challenges of ageing. Nature 561:45–56. https://doi.org/10.1038/s41586-018-0457-8
Paulsen G et al (2014) Vitamin C and E supplementation hampers cellular adaptation to endurance training in humans: a double-blind, randomised, controlled trial. J Physiol 592:1887–1901. https://doi.org/10.1113/jphysiol.2013.267419
Perez-de-Arce K, Foncea R, Leighton F (2005) Reactive oxygen species mediates homocysteine-induced mitochondrial biogenesis in human endothelial cells: modulation by antioxidants . Biochem Biophys Res Commun 338:1103–1109. https://doi.org/10.1016/j.bbrc.2005.10.053
Puigserver P, Spiegelman BM (2003) Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev 24:78–90. https://doi.org/10.1210/er.2002-0012
Radak Z, Zhao Z, Koltai E, Ohno H, Atalay M (2013) Oxygen consumption and usage during physical exercise: the balance between oxidative stress and ROS-dependent adaptive signaling. Antioxid Redox Signal 18:1208–1246. https://doi.org/10.1089/ars.2011.4498
Reczek CR, Chandel NS (2015) ROS-dependent signal transduction . Curr Opin Cell Biol 33:8–13. https://doi.org/10.1016/j.ceb.2014.09.010
Ristow M et al (2009) Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci USA 106:8665–8670. https://doi.org/10.1073/pnas.0903485106
Romanello V, Sandri M (2015) Mitochondrial quality control and muscle mass maintenance. Front Physiol 6:422. https://doi.org/10.3389/fphys.2015.00422
Romanello V, Scalabrin M, Albiero M, Blaauw B, Scorrano L, Sandri M (2019) Inhibition of the fission machinery mitigates OPA1 impairment in adult skeletal muscles. Cells. https://doi.org/10.3390/cells8060597
Safdar A, Little JP, Stokl AJ, Hettinga BP, Akhtar M, Tarnopolsky MA (2011) Exercise increases mitochondrial PGC-1alpha content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. J Biol Chem 286:10605–10617. https://doi.org/10.1074/jbc.M110.211466
Sakellariou GK, Vasilaki A, Palomero J, Kayani A, Zibrik L, McArdle A, Jackson MJ (2013) Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid Redox Signal 18:603–621. https://doi.org/10.1089/ars.2012.4623
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141:290–303. https://doi.org/10.1016/j.cell.2010.02.024
Schiavi A, Ventura N (2014) The interplay between mitochondria and autophagy and its role in the aging process. Exp Gerontol 56:147–153. https://doi.org/10.1016/j.exger.2014.02.015
Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM (1998) A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol 143:351–358. https://doi.org/10.1083/jcb.143.2.351
Sobotta MC et al (2015) Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat Chem Biol 11:64–70. https://doi.org/10.1038/nchembio.1695
Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC (2009) Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell 20:3525–3532. https://doi.org/10.1091/mbc.E09-03-0252
St-Jean-Pelletier F et al (2017) The impact of ageing, physical activity, and pre-frailty on skeletal muscle phenotype, mitochondrial content, and intramyocellular lipids in men. J Cachexia Sarcopenia Muscle 8:213–228. https://doi.org/10.1002/jcsm.12139
Stocker S, Maurer M, Ruppert T, Dick TP (2018) A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat Chem Biol 14:148–155. https://doi.org/10.1038/nchembio.2536
Tezze C et al (2017) Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab 25:1374-1389e.6. https://doi.org/10.1016/j.cmet.2017.04.021
Twig G et al (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27:433–446. https://doi.org/10.1038/sj.emboj.7601963
Valenzuela PL, Castillo-Garcia A, Morales JS, Izquierdo M, Serra-Rexach JA, Santos-Lozano A, Lucia A (2019) Physical exercise in the oldest old. Compr Physiol 9:1281–1304. https://doi.org/10.1002/cphy.c190002
Vasilaki A, McArdle F, Iwanejko LM, McArdle A (2006) Adaptive responses of mouse skeletal muscle to contractile activity: the effect of age. Mech Ageing Dev 127:830–839. https://doi.org/10.1016/j.mad.2006.08.004
Vega RB, Huss JM, Kelly DP (2000) The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol 20:1868–1876. https://doi.org/10.1128/mcb.20.5.1868-1876.2000
Winterbourn CC (2015) Are free radicals involved in thiol-based redox signaling? Free Radic Biol Med 80:164–170. https://doi.org/10.1016/j.freeradbiomed.2014.08.017
Wu Z et al (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98:115–124. https://doi.org/10.1016/S0092-8674(00)80611-X
Yamada M, Iwata M, Warabi E, Oishi H, Lira VA, Okutsu M (2019) p62/SQSTM1 and Nrf2 are essential for exercise-mediated enhancement of antioxidant protein expression in oxidative muscle. FASEB J 33:8022–8032. https://doi.org/10.1096/fj.201900133R
Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion and stress. Science 337:1062–1065. https://doi.org/10.1126/science.1219855
Yu X, Long YC (2015) Autophagy modulates amino acid signaling network in myotubes: differential effects on mTORC1 pathway and theintegrated stress response. FASEB J 29(2):394–407. https://doi.org/10.1096/fj.14-252841
Zhang Y, Chan NC, Ngo HB, Gristick H, Chan DC (2012) Crystal structure of mitochondrial fission complex reveals scaffolding function for mitochondrial division 1 (Mdv1) coiled coil. J Biol Chem 287:9855–9861. https://doi.org/10.1074/jbc.M111.329359
Zhang L, Zhang Z, Khan A, Zheng H, Yuan C, Jiang H (2020) Advances in drug therapy for mitochondrial diseases. Ann Transl Med 8:17. https://doi.org/10.21037/atm.2019.10.113
Zinovkina LA, Zinovkin RA (2015) DNA methylation, mitochondria, and programmed aging . Biochemistry (Mosc) 80:1571–1577. https://doi.org/10.1134/S0006297915120044
Acknowledgements
A.S. was a physiology student at NUIG. The authors wish to thank the Discipline of Physiology, School of Medicine, NUI Galway for support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shally, A., McDonagh, B. The redox environment and mitochondrial dysfunction in age-related skeletal muscle atrophy. Biogerontology 21, 461–473 (2020). https://doi.org/10.1007/s10522-020-09879-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10522-020-09879-7