
Abstract
Age-related bone loss has been associated with high levels of marrow adipogenesis. Estrogens (E2) are known to regulate the differentiation of marrow precursors into osteoblasts, however, their role in bone marrow adipogenesis remain unknown. E2 regulate adipocyte differentiation in subcutaneous and visceral fat through interaction with other nuclear receptors. This interaction has not been assessed in bone marrow adipocytes in vivo. In this study, we compared two groups of animals, young and old, after either oophorectomy (OVX) or oophorectomy plus E2 (OVX + E2) replacement. We found that absence of E2 was associated with higher levels of PPARγ and lower levels of Sirt1 most significantly in the old group. In addition, old mice responded better to E2 replacement in terms of reducing adipogenesis and PPARγ expression as well as increasing levels of Sirt1 expression. Our findings represent a new understanding of the role of E2 in age-related bone loss, which could be mediated through the regulation of Sirt1 expression within the bone marrow. In addition, this evidence suggests that old individuals may show a better response to E2 administration in terms of reverting the high levels of marrow fat seen in age-related bone loss.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Estrogens (E2) increase bone mass through the regulation of osteoclastic activity (Chaiamnuay and Saag 2006). Osteoclast progenitors respond to E2 through their conventional E2 receptors by decreasing osteoclast survival and activity (Zallone 2006). During the menopause, a reduction in serum E2 is followed by a subsequent increase of osteoclastic activity and bone loss. This is considered the main pathophysiological mechanism of post-menopausal osteoporosis (Lund 2008; Raisz and Seeman 2001). In contrast to post-menopausal osteoporosis, age-related bone loss is due not only to high levels of resorption, but also to increasing levels of bone marrow adipogenesis at expense of osteoblastogenesis, thus decreasing bone formation (Duque and Troen 2008).
In vivo studies, both in rats (Sottile et al. 2004) and humans (Syed et al. 2008), have demonstrated that E2 deprivation enhances marrow fat infiltration by increasing bone marrow adipocyte number and size, which was reverted by E2 replacement. However, the mechanisms explaining these findings have not been elucidated.
Among the proposed mechanisms of increasing adipogenesis in age-related bone loss, high levels of expression of the nuclear receptor peroxisome proliferator activator gamma (PPARγ) is the most widely accepted and studied (Duque and Troen 2008; Shockley et al. 2007). Pharmacological induction of PPARγ inhibits osteoblast differentiation while favouring adipogenic conversion of precursor cells (Moerman et al. 2004). In contrast, downregulation of PPARγ in vivo is associated with higher bone mass (Akune et al. 2004).
Considering that both, PPARγ and E2 receptors, belong to the nuclear binding receptors and share similar coactivators (Kumar and Thompson 1999), the effect of E2 on adipogenesis could be associated with their interaction with PPARγ in precursor cells. In fact, several studies have suggested an inhibitory effect of E2 on PPARγ (Foryst-Ludwig et al. 2008), therefore reducing adipogenesis. This effect has been tested using several types of E2 and E2-like compounds (Foryst-Ludwig et al. 2008; Liao et al. 2008).
Among these compounds, resveratrol is a phytoestrogen with both a potent inhibitory effect on adipogenesis (Rayalam et al. 2008), and a stimulatory effect on osteoblastogenesis (Bäckesjö et al. 2006). The effect of resveratrol on mesenchymal stem cells differentiation is exerted through the activation of the nuclear NAD-dependent protein deacetylase Sirt1 (Bäckesjö et al. 2006). Interestingly, Sirt1 activation represses the action of PPARγ and adipocyte differentiation in subcutaneous and visceral fat (Picard et al. 2004). However, the role of Sirt1 in bone marrow adipogenesis in vivo remains unknown.
Since most of the studies looking at the potential association between nuclear receptors, E2 and sirtuins have been performed in subcutaneous and visceral fat in vitro, in this in vivo study, we compared two groups of animals, young and old, and analyzed the changes in the expression of PPARγ and Sirt1 in bone marrow fat after either oophorectomy (OVX) or oophorectomy plus E2 (OVX + E2) replacement. We found that absence of E2 was associated with higher levels of PPARγ and lower levels of Sirt1, most significantly in the old group. In addition, old mice responded better to E2 replacement in terms of reducing adipogenesis and PPARγ expression, as well as increasing levels of Sirt1 expression.
Our findings represent a new understanding of the role of E2 in age-related bone loss, which could be mediated through the regulation of Sirt1 expression within the bone marrow. In addition, this evidence suggests that old individuals may show a better response to E2 administration in terms of reverting the high levels of marrow fat seen in age-related bone loss.
Methods
Animal tissue preparation
All protocols were approved by the McGill University Animal Care Utilization Committee and carried out in accordance with the requirements of the Canadian Council on Animal Care. Mice were kindly provided by Dr. Marilyn Miller from the Department of Gynecology and Obstetrics, McGill University. Young skeletally mature (5 months) and old (22–24 months) female C57BL/6J mice were either gonadally intact, OVX or OVX + E2 (estratien-3,17-β-diol Sterloids Inc., Witten, NH, USA). Mice were maintained in an isolated, pathogen-free colony with a 12-h light/dark photoperiod and food and water ad libitum. In normal animals, reproductive cyclicity patterns were established by daily vaginal smears. Young intact mice showed all stages of the reproductive cycle and old intact mice were in persistent diestrus. Surgery was performed during diestrus and tissue collected as previously described (Duque et al. 2002). Under anesthesia with ketamine (8.5 mg/100 g) and xylazine (0.3 mg/100 g) i.m, bilateral OVX surgery was performed using a posterior surgical approach and 5 mm silastic capsules containing E2 crystals mixed in silastic adhesive were then implanted subcutaneously (s.c.) in the nape of the neck for OVX + E2 animals. These capsules produce mean circulating levels of E2 of 12–18 ng/ml, i.e., typical of diestrus animals, whereas levels of estradiol for OVX animals were undetectable. The OVX + E2 mice received treatment with E2 for 6 weeks. For the duration of the E2 treatment, control mice (intact and OVX) were kept in the same conditions as OVX + E2 mice. The completeness of gonadectomy and efficiency of E2 treatment was determined by the appearance of vaginal cytological smears exhibiting the absence or presence of mature epithelial cells, respectively. Post-mortem uterus weight was taken as an index of the effect of E2 deprivation (OVX) or replacement (E2 treatment), and was significantly reduced (P < 0.05) in OVX group (44 ± 1 mg, mean ± SEM) compared to intact (76 ± 2 mg) or OVX + E2 mice (64 ± 3 mg).
Six treatment groups were evaluated. (a) Normally cycling (young—5 months, n = 6; old—24 months, n = 6; intact): (b) OVX (young—5 months, n = 6; old—24 months, n = 6): (c) OVX treated with 17 β Estradiol (young—5 months, n = 6; old—24 months, n = 6). Animals were sacrificed by decapitation. Femur was placed in 4% paraformaldehyde for further histology analysis. Tibiae were isolated for bone marrow flushing and protein extraction.
Decalcification of tissues
EDTA-glycerol (EDTA-G) solution was prepared as previously described (Duque et al. 2002). 14.5 g EDTA, 1.25 g NaOH, and 15 ml glycerol were dissolved in distilled water and the pH was adjusted to 7.3. The solution was then made up to 100 ml and stored at 5°C.
After fixation, specimens were serially washed for 12 h at 5°C in each of the following solutions: 0.01 M PBS containing 5% glycerol, 0.01 M PBS containing 10% glycerol, and 0.01 M PBS containing 15% glycerol. The specimens were then decalcified in EDTA-G solution for 10–14 days at 5°C. This EDTA-G solution was replaced every 5 days. Progression of decalcification was checked by X-ray. To remove EDTA and glycerol from the decalcified tissues, they were washed at 5°C for 12 h successively in 15% sucrose and 15% glycerol in PBS; 20% sucrose and 10% glycerol in PBS; 20% sucrose and 5% glycerol in PBS; 20% sucrose in PBS; 10% sucrose in PBS; 5% sucrose in PBS and 100% PBS as previously described (Duque et al. 2002).
Adipocyte quantification
For adipocyte quantification, the right femur from 6 mice per group was cleaned of soft tissue, fixed for 36 h in 4% paraformaldehyde, rinsed thoroughly in PBS, decalcified, and processed for paraffin embedding. Serial 4-μm sections (5 per mouse) cut on a modified Leica RM 2155 rotary microtome (Leica Microsystems, Richmond Hill, Ontario, Canada) were deparaffinized in xylene and immersed in two changes of propylene glycol before staining with haematoxylin/eosin to quantify adipocyte number as previously described (Duque et al. 2004a). Adipocyte number was measured by tracing out individual adipocytes in all the fields analyzed (10 per section); adipocytes appear as distinct, translucent, yellow ellipsoids in the marrow cavity. All measurements were done at a 40× magnification using the Bioquant image analysis software (Bioquant Corp. Nashville, TN, USA.) Photomicrographs were captured at a 40× magnification.
Detection of PPARγ and Sirt1 by immunofluorescence
After decalcification, bone samples were embedded in low-melting-point paraffin in a Shandon Citadel 2000 automatic tissue processor (Shandon Scientific Limited, Runcorn, UK). Coronary and transverse consecutive serial sections (4 μ) were made for the epiphyseal parts and the shaft, respectively. Sections were mounted on silane-coated glass slides (Fischer Scientific, Springfield, NJ, USA) and paraffin was removed with three washes of xylene and rehydrated with ethanol washes (80–50–30%) and PBS. Non-specific binding was blocked by addition of goat serum for 1 h. Sections were then incubated with either goat polyclonal IgG PPARγ or mouse monoclonal Sirt1 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) with 1.5% normal blocking serum overnight at 4°C and then incubated for 45 min with either phytoerythrin-conjugated mouse for PPARγ or FITC-conjugated donkey anti mouse for Sirt1 (Santa Cruz, Santa Cruz, CA, USA) diluted to 2 μg/ml in PBS with 1.5% normal blocking serum. In all immunofluorescence detection methods for PPARγ and Sirt1, non-immune mouse serum and mouse IgG, in place of the primary antibody, were included as controls.
Quantification of fluorescence staining
Bioquant image analysis was used to analyze fluorescent images. Threshold intensities for each picture were determined and from this the number of pixels within the picture that were above the threshold intensity were calculated and considered positively stained. The positively stained pixels were expressed as a percentage of the total pixels in the picture.
Western blot analysis
For western blot analysis, the bone marrow was flushed from the left tibiae, and resuspended in SDS-sample buffer for total protein extraction. 20 μg of total protein were loaded on polyacrylamide gels and separated by standard SDS-polyacrylamide gel electrophoresis. To control for variances in gel migration, exposure time, antibody incubation etc., samples were run on the same gels and transferred to the same PVDF membranes (Amersham). Blots were blocked overnight in 2.5% non-fat dried milk and probed with antibodies directed against Sirt1 and PPARγ2 (1:1,000, Santa Cruz Biotechnology, Santacruz, CA, USA). Positive controls were included in all experiments as provided by the manufacturer (Santa Cruz Biotechnology, Santacruz, CA, USA, USA), to confirm antibody specificity. Secondary antibodies conjugated to horseradish peroxidase were from Sigma (1:5,000). Antigen-antibody complexes were detected by chemiluminescence using a kit of reagents from ECL (Amersham, UK) and blots were exposed to high-performance chemiluminescence film (Amersham, UK). Films were scanned and the optical density of each specific band analyzed using the ImageMaster program and expressed as OD/mm2/100 μg of total protein. Relative intensity of the samples was determined by comparing the protein of interest versus control (tubulin). Values are reported as the average of samples obtained from six mice.
Statistical analysis
Results are expressed as means ± SD and differences between groups of mice were determined by the Student t test and the Mann–Whitney test. A P value of ≤0.05 was considered statistically significant.
Results
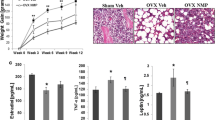
Intact old mice showed a significantly higher adipocyte number compared to the intact young mice (25 ± 2 vs. 8 ± 2, P < 0.01; Fig. 1). In both young and old mice, OVX induced higher levels of adipocytes numbers as compared with their intact controls (P < 0.01). This increase was significantly higher in old OVX mice as compared to young OVX mice (45 ± 3 vs. 18 ± 1, P < 0.001). In both young and old OVX mice, E2 replacement induced a significant reduction in adipocyte number (P < 0.01). This reduction was significantly higher in the old OVX + E2 group (P < 0.001).

Changes in bone marrow fat and adipocyte number in young and old OVX C57BL/6J mice. Sections of undecalcified bone were stained with H/E and images captured at original magnifications of ×40 to evaluate fat infiltration (a, white areas, red arrows) and adipocyte number per field (b). Both, young and old intact mice, showed infiltration of bone marrow by fat with more significant infiltration happening in the old group. In addition, OVX significantly increased marrow fat infiltration in both young and old mice. This increase in marrow fat was normalized by E2 replacement. Magnification at source was ×40. Micrographs are representative of four to six screened in each group of animals. Average of adipocyte number was quantified in ten fields per section. Bars 50 μm. * P < 0.01 compared with young intact mice; ** P < 0.001 compared with young intact mice, δP < 0.001 compared with old intact mice
PPARγ expression was significantly lower in all young mice groups compared to the old mice groups (Fig. 2a). Quantification of PPARγ expression by immunofluorescence showed a significant increase in bone marrow expression after OVX in both young and old mice (Fig. 2, P < 0.01). This increase in PPARγ expression after OVX was significantly higher in old OVX mice (P < 0.001). In both groups, young and old OVX, PPARγ expression returned to baseline after E2 replacement in the corresponding age group.

Changes in PPARγ expression in bone marrow of young and old OVX C57BL/6J mice. After decalcification, bone samples were embedded in low-melting paraffin, coronary sections were made for the epiphyseal parts and the shaft, respectively. Sections were treated as described in materials and methods. In both cases sections were incubated with PPARγ2 antibody (red fluorescence). Each longitudinal section was analyzed using Bioquant image analysis. The positively staining pixels were expressed as a percentage of the total pixels in the picture (b). PPARγ expression was lower in all young mice groups as compared with the old mice groups (a). Quantification of PPARγ expression by immunofluorescence showed a significant increase in bone marrow expression after OVX in both young and old mice returning to normal levels after E2 replacement. In addition, old OVX showed a significantly higher increase in PPARγ expression as compared with the young OVX mice. Bars 50 μm. * P < 0.01 compared with young intact mice; ** P < 0.001 compared with young intact mice, δP < 0.001 compared with old intact mice
Furthermore, in contrast with PPARγ expression, OVX induced a significant reduction in Sirt1 expression in both young and old mice (Fig. 3, P < 0.01). In addition, young intact mice showed higher levels of Sirt1 than old intact mice (Fig. 3, P < 0.01). Expression levels of Sirt1 in young and old OVX were brought to baseline after E2 replacement. E2 replacement induced a significantly higher increase in Sirt1 protein expression in bone marrow of old OVX mice as compared with E2-replaced young OVX mice (P < 0.001).

Changes in Sirt1 expression in bone marrow of young and old OVX C57BL/6J mice. After decalcification, bone samples were embedded in low-melting paraffin, coronary sections were made for the epiphyseal parts and the shaft, respectively. Sections were treated as described in materials and methods. In both cases sections were incubated with Sirt1 antibody (green fluorescence). Each longitudinal section was analyzed using Bioquant image analysis. The positively staining pixels were expressed as a percentage of the total pixels in the picture (b). Sirt1 expression was higher in young intact mice groups as compared with the old intact group (a). Quantification of Sirt1 expression by immunofluorescence showed a significant decrease in bone marrow expression after OVX in both young and old mice, returning to normal levels after E2 replacement. In addition, old OVX showed a significantly higher increase in Sirt1 expression after E2 replacement as compared with the young mice. Bars 50 μm. * P < 0.01 compared with young intact mice; ** P < 0.001 compared with young intact mice, δP < 0.001 compared with old intact mice
Finally, immunofluorescence quantification of PPARγ and Sirt1 expression were confirmed by western blot using protein extracts from bone marrow cells (Fig. 4). In agreement with immunofluorescence data, E2 replacement in old mice induced significantly lower levels of PPARγ and a significantly higher increase in Sirt1 protein expression in bone marrow of old OVX mice as compared with E2-replaced young OVX mice (P < 0.01).

Western blot analysis of PPARγ and Sirt1 expression in bone marrow of young and old OVX C57BL/6J mice. Western blot analysis of total protein obtained from bone marrow flushing in young and old OVX C57BL/6J mice treated with E2. In agreement with immunofluorescence data (Figs. 2, 3), E2 replacement in old mice induced significantly lower levels of PPARγ and a significantly higher increase in Sirt1 protein expression in bone marrow of old OVX mice as compared with E2-replaced young OVX mice. Data from scanning densitometric analyses is expressed as the ratio between the protein of interest and tubulin as control representing the mean ± SD of triplicate determinations. * P < 0.01; δP < 0.001 for old groups versus their corresponding young controls
Discussion
In this study, changes in PPARγ and Sirt1 expression were compared after either OVX or OVX + E2 replacement in young vs. old C57BL/6J mice. Our data indicate that OVX induced a significant increase in bone marrow fat in old mice compared to young mice. In addition, the increase in marrow fat in OVX old mice correlated with significantly higher levels of PPARγ expression within the bone marrow, which were normalized after E2 replacement. Furthermore, Sirt2 levels within the bone marrow were significantly reduced after OVX in both young and old mice. Finally, old OVX mice showed a significantly higher increase in Sirt1 expression after E2 replacement as compared with OVX + E2 young mice.
The increasing levels of bone marrow adipogenesis in age-related bone loss have been correlated with high levels of PPARγ expression by marrow cells (Duque et al. 2004b). The mechanisms that explain this age-related increase in PPARγ are subject of intense research. Levels of ligands for PPARγ such as polyunsaturated fatty acids and eicosanoids, are known to increase with aging (Moerman et al. 2004). In the case of E2, it is known that E2 act as modulators of the marrow stromal precursor differentiation and therefore constitutes an important determinant factor in balancing osteoblastogenesis over adipogenesis (Hong et al. 2006).
Previous studies in young OVX rats (6 and 9 month-old) have demonstrated an increase in bone marrow adipogenesis (Sottile et al. 2004). However, the effect of OVX and E2 on marrow adipogenesis has not been assessed in an accepted mouse model of age-related bone loss, therefore in this study we compared young (5 months) vs. very old (24 months) C57BL/6J mice. The age-related changes in bone phenotype of these mice have been extensively studied (Ferguson et al. 2003), including their changes in bone mass and calcium metabolism induced by OVX (Kalu and Chen 1999). In agreement with previous studies in young rats (Sottile et al. 2004), our findings in young and old mice suggest that E2 replacement reverses adipogenesis to baseline.
In an attempt to elucidate a potential mechanism that could partially explain the role of E2 deficiency in increasing bone marrow adipogenesis with age, we looked at changes in Sirt1 expression within the bone marrow induced by either OVX or OVX + E2. Considering that Sirt1 is a known inhibitor of PPARγ in adipocytes obtained from subcutaneous and visceral fat (Picard et al. 2004), and that Sirt1 expression is increased by the presence of E2 in in vitro models (Rasbach and Schnellmann 2008), it is tempting to hypothesize that high levels of PPARγ after OVX would correlate with low levels of Sirt2, which would be corrected by E2 replacement.
In our model, OVX induced both high levels of marrow adipogenesis and PPARγ expression in bone marrow of both young and old mice. However, this increase was significantly higher in the old mice. A particularly interesting finding of our study is that E2 replacement had a more significant effect in the old than the young animals suggesting that old individuals who receive E2 would benefit more than young individuals in terms of inhibition of marrow adipogenesis. In fact, a recent study by Syed et al. (2008) in female subjects (mean age 63 year-old) treated with E2 reported a significant decrease in marrow adipogenesis. According with our findings, older subjects would benefit more of E2 replacement. Further studies looking at this effect in older populations should be pursued.
Although our data is unable to establish a direct causal relationship between high levels of PPARγ and low levels of Sirt1 after OVX, we provide evidence correlating the changes seen in Sirt1 with those seen in PPARγ expression after OVX and E2 replacement. In addition, the significant recovery in Sirt1 expression after E2 replacement in old mice suggests that E2 replacement has a positive stimulatory effect on Sirt1 expression in bone. The significance of this finding should be studied in the future.
In summary, using a model of OVX and E2 replaced aging mice we have found that increasing levels of adipogenesis within the bone marrow after OVX could be associated with high levels of PPARγ and low levels of Sirt1 expression. High levels of marrow adipogenesis were normalized by E2 replacement with a significantly higher response in the old mice.
In conclusion, E2 play an important role in the pathogenesis and treatment of age-related bone loss. The understanding of the mechanisms of action of E2 on bone marrow cellularity would provide us with new potential therapeutic approach to promote osteoblastogenesis at the expense of adipogenesis and thus increase bone mass and prevent osteoporosis in our aging population.
References
Akune T, Ohba S, Kamekura S et al (2004) PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest 113:846–855
Bäckesjö CM, Li Y, Lindgren U et al (2006) Activation of Sirt1 decreases adipocyte formation during osteoblast differentiation of mesenchymal stem cells. J Bone Miner Res 21:993–1002. doi:10.1359/jbmr.060415
Chaiamnuay S, Saag KG (2006) Postmenopausal osteoporosis. What have we learned since the introduction of bisphosphonates? Rev Endocr Metab Disord 7:101–112. doi:10.1007/s11154-006-9008-y
Duque G, Troen BR (2008) Understanding the mechanisms of senile osteoporosis: new facts for a major geriatric syndrome. J Am Geriatr Soc 56:935–941. doi:10.1111/j.1532-5415.2008.01764.x
Duque G, El Abdaimi K, Macoritto M et al (2002) Estrogens (E2) regulate expression and response of 1, 25-dihydroxyvitamin D3 receptors in bone cells: changes with aging and hormone deprivation. Biochem Biophys Res Commun 299:446–454. doi:10.1016/S0006-291X(02)02657-8
Duque G, Macoritto M, Kremer R (2004a) Vitamin D treatment of senescence accelerated mice (SAM-P/6) induces several regulators of stromal cell plasticity. Biogerontology 5:421–429. doi:10.1007/s10522-004-3192-5
Duque G, Macoritto M, Kremer R (2004b) 1,25(OH)2D3 inhibits bone marrow adipogenesis in senescence accelerated mice (SAM-P/6) by decreasing the expression of peroxisome proliferator-activated receptor gamma 2 (PPARgamma2). Exp Gerontol 39:333–338. doi:10.1016/j.exger.2003.11.008
Ferguson VL, Ayers RA, Bateman TA et al (2003) Bone development and age-related bone loss in male C57BL/6J mice. Bone 33(3):387–389. doi:10.1016/S8756-3282(03)00199-6
Foryst-Ludwig A, Clemenz M, Hohmann S et al (2008) Metabolic actions of estrogen receptor beta (ERbeta) are mediated by a negative cross-talk with PPARgamma. PLoS Genet 4:e1000108. doi:10.1371/journal.pgen.1000108
Hong L, Colpan A, Peptan IA (2006) Modulations of 17-beta estradiol on osteogenic and adipogenic differentiations of human mesenchymal stem cells. Tissue Eng 12:2747–2753. doi:10.1089/ten.2006.12.2747
Kalu DN, Chen C (1999) Ovariectomized murine model of postmenopausal calcium malabsorption. J Bone Miner Res 14:593–601. doi:10.1359/jbmr.1999.14.4.593
Kumar R, Thompson EB (1999) The structure of the nuclear hormone receptors. Steroids 64:310–319. doi:10.1016/S0039-128X(99)00014-8
Liao QC, Li YL, Qin YF et al (2008) Inhibition of adipocyte differentiation by phytoestrogen genistein through a potential downregulation of extracellular signal-regulated kinases 1/2 activity. J Cell Biochem 104:1853–1864. doi:10.1002/jcb.21753
Lund KJ (2008) Menopause and the menopausal transition. Med Clin North Am 92:1253–1271. doi:10.1016/j.mcna.2008.04.009
Moerman EJ, Teng K, Lipschitz DA et al (2004) Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell 3:379–389. doi:10.1111/j.1474-9728.2004.00127.x
Picard F, Kurtev M, Chung N et al (2004) Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 429:771–776. doi:10.1038/nature02583
Raisz LG, Seeman E (2001) Causes of age-related bone loss and bone fragility: an alternative view. J Bone Miner Res 16:1948–1952. doi:10.1359/jbmr.2001.16.11.1948
Rasbach KA, Schnellmann RG (2008) Isoflavones promote mitochondrial biogenesis. J Pharmacol Exp Ther 325:536–543. doi:10.1124/jpet.107.134882
Rayalam S, Yang JY, Ambati S et al (2008) Resveratrol induces apoptosis and inhibits adipogenesis in 3T3–L1 adipocytes. Phytother Res 22:1367–1371. doi:10.1002/ptr.2503
Shockley KR, Rosen CJ, Churchill GA et al (2007) PPARgamma2 regulates a molecular signature of marrow mesenchymal stem cells. PPAR Res 2007:81219. doi:10.1155/2007/81219
Sottile V, Seuwen K, Kneissel M (2004) Enhanced marrow adipogenesis and bone resorption in estrogen-deprived rats treated with the PPARgamma agonist BRL49653 (rosiglitazone). Calcif Tissue Int 75:329–337. doi:10.1007/s00223-004-0224-8
Syed FA, Oursler MJ, Hefferanm TE et al (2008) Effects of estrogen therapy on bone marrow adipocytes in postmenopausal osteoporotic women. Osteoporos Int 19:1323–1330. doi:10.1007/s00198-008-0574-6
Zallone A (2006) Direct and indirect estrogen actions on osteoblasts and osteoclasts. Ann N Y Acad Sci 1068:173–179. doi:10.1196/annals.1346.019
Acknowledgments
This work was supported by grants from the Canadian Institutes of Health Research and the Nepean Medical Research Foundation. Dr. Duque holds a Fellowship from the University of Sydney-Medical Research Foundation. The authors would like to thank Mrs. Leigh Bambury for preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Elbaz, A., Rivas, D. & Duque, G. Effect of estrogens on bone marrow adipogenesis and Sirt1 in aging C57BL/6J mice. Biogerontology 10, 747–755 (2009). https://doi.org/10.1007/s10522-009-9221-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10522-009-9221-7