
Abstract
Genes that influence a phenotype earlier in life may differ from those influencing the same phenotype later, particularly during significant development periods such as puberty, when it is known that new genetic and environmental influences may become important. In the present study, body mass index (BMI) data were collected from 470 monozygotic twin pairs and 673 dizygotic twin pairs longitudinally at ages 12, 14 and 16, roughly straddling puberty. In order to examine whether there are qualitative and quantitative differences in genetic and environmental influences affecting BMI in males and females, during development, a general sex-limitation simplex model (which represents the longitudinal time series of the data) was fitted to the repeated measurements of BMI. The ADE simplex model provided the best fit to the adolescent data, with disparity in the magnitude of additive genetic influences between sexes, but no differences in the non-additive genetic (epistasis or dominance) or environmental influences. Results found may reflect many genetic and environmental influences during puberty, including the possible complex interaction between genes involved in the biological mechanism of weight regulation and the development of likely peer pressured activities such as severe exercise and diet regimes. Although, over 1,000 pairs of twins were used, this study still lacked the power to properly discriminate between additive and non-additive genetic variance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Many studies have investigated the role of genes and environment in the variability of relative weight, as defined by body mass index (BMI). BMI is an anthropometric measure of body mass according to height and is one of the most extensively studied obesity-related phenotypes. Obesity causes or exacerbates many health problems such as type II diabetes mellitus, coronary heart disease, certain forms of cancer, respiratory complications and osteoarthritis. The worldwide prevalence of being overweight appears to be rising not only in adults but also in children (Chambers 2001). In Australia, almost a quarter of children and adolescents are overweight or obese (Australian Institute of Health and Welfare (AIHW) 2000; Eckersley 2003). In fact, the incidence of obesity induced type II diabetes among children increased tenfold between 1982 and 1994 (Hill et al. 2003).
Body weight is a multifactorial trait, determined by an interaction between environmental and genetic factors. Indisputably, obesity has a significant heritable contribution. However, estimates of genetic and environmental effects vary widely, depending on study type, population and age of subjects. The few studies performed in children, adolescents and young adults show heritabilities for BMI from 70% to over 90% (Bouchard et al. 1985; Allison et al. 1994a; Maes et al. 1997; Pietilainen et al. 1999). In adult twin populations, variability of body weight ascribed to genes is between 50 and 80% (Pietilainen et al. 1999). Adoption studies show only moderate effects of genes (30–40%) while family studies generally yield estimates of heritability intermediate between twin and adoption studies (Bouchard et al. 1985; Pietilainen et al. 1999).
The consistent differences in these estimates may suggest that, in part, the heritable variance is due to non-additive genetic effects, that is, dominance and recessivity and/or non-allelic gene interaction (epistasis). Studies in several populations suggest that major genes inherited in a recessive manner may account for 35–45% of the variation in obesity-related traits (Borecki et al. 1998; Perola et al. 2001; Deng et al. 2002). On the other hand, twin studies in Australian and European adult population cohorts have found that the variability of BMI is mainly influenced by additive genetic and unique environmental sources.(Schousboe et al. 2003; Cornes et al. 2005). Thus, shared environmental effects appear to play little, if any, role in BMI variance, although it is possible that common environment exerts a stronger influence on weight during adolescence, when siblings reared together are experiencing shared environment directly.
Little is known about the developmental changes in genetic and environmental influences on body weight, especially during a major transition such as puberty when genes play an important role in the biological mechanism of weight regulation. In the past, very few longitudinal studies of twins have been conducted in paediatric samples to address the heritability of BMI. Results from longitudinal studies in adult samples support the suggestions from cross-sectional data that there are age-specific genetic effects on BMI (Neale and Cardon 1992; Meyer 1995). This can be particularly true during puberty, when complex sequences of hormones are involved in human adolescent growth and maturation. Concerted actions of growth hormones and sex and adrenal steroids are responsible for timing of growth spurts, stabilization of the growth processes and attainment of maturity (Loesch et al. 1995; Roemmich and Rogol 1999). However, a disadvantage of cross-sectional data analyses is that they do not allow for the discrimination of those transient factors that may exert a long term influence from one time point to another.
Another area of ambiguity concerns sex-limitation in the heritability of adiposity. There are well known gender specific differences in weight from birth to adulthood. Prenatally and at subsequent ages, girls are physiologically more mature than boys. There is also evidence to suggest that genetic effects contributing to the aetiology of variation in BMI are different in males and females. In a sample of Finnish teenagers, different sets of genes accounted for the additive genetic variation in BMI in males and females (Pietilainen et al. 1999). In contrast, in two studies of younger twins, no gender effects for BMI were found in either adolescents (Allison et al. 1994a) or in a pre-adolescent population (Bodurtha et al. 1990). In addition, total heritability of BMI or body fat in adults seems to be different depending on gender, although studies have yielded equivocal findings. Some studies have found that females have a higher heritability for BMI than males (Neale and Cardon 1992; Allison et al. 1994b; Harris et al. 1995) including an Australian population study of 933 adult twin families (Cornes et al. 2005). Others report the opposite (Stunkard et al. 1990; Korkeila et al. 1991). A Swedish adoption/twin data study reported a higher heritability of BMI for men, but the primary sex difference was the relatively large contribution of non-additive genetic effects among men (Stunkard et al. 1990; Harris et al. 1995).
To summarise, the mode of inheritance and the underlying complexity of obesity remain largely unresolved. Moreover, while previous model-fitting analyses are consistent in implicating genetic influences in BMI, they are inconsistent regarding whether those influences are additive and/or non-additive. Furthermore, the presence of distinct male and female patterns of weight gain and fat distribution suggest that the genetic and environmental effects contributing to the variation in body mass may not be identical, or may operate differently, in males and females throughout development. Finally, the majority of studies conducted in paediatric samples use cross-sectional data, which only allows for the discrimination of factors that affect measurement at one time point.
In light of these factors, we have measured BMI longitudinally at ages 12, 14 and 16 in 1,143 monozygotic (MZ) and dizygotic (DZ) twin pairs. By performing sex-limited longitudinal analysis on the adolescent twin data, we were able to establish whether, during puberty, genetic and environmental determinants of relative weight are the same in males and females. More specifically: (a) do genetic and environmental influences for BMI change in magnitude between different stages of puberty? (b) do the same genetic and environmental sources occur at different times during puberty? and (c) are the genetic and environmental influences on BMI similar in males and females? More importantly, our results from these analyses may add to the current limited knowledge of the genes regulating body weight during puberty.
Methods
Subjects and measures
MZ and DZ twin pairs were measured longitudinally for height and weight at the ages of 12, 14 and 16. At ages 12 and 14, data were collected as part of an ongoing study on melanoma risk factors (Zhu et al. 1999). At age 16, data were collected in the context of an ongoing study of cognition (Wright et al. 2001) in which most twins had also taken part in the melanoma risk factors study. Twins involved in both studies were recruited by contacting principals of primary schools in the greater Brisbane area, through word of mouth and by media appeals. The twins who registered their interest were contacted and participation was conditional upon the informed consent of the twins and their parents. In this study, we report on data collected from January 1992 until April 2006.
Data were collected from 1,143 complete twin pairs, 470 MZ, 346 DZ same-sex (DZSS) and 327 DZ opposite sex (DZOS) twin pairs. Data were also collected from 570 non twin siblings but are not analysed here. Height and weight were both measured clinically using a stadiometer and accurate scales, respectively. BMI was calculated by weight (in kilograms)/ height (in metres)2. To minimise age bias, measurements were taken as close as possible to the twins’ 12th, 14th and 16th birthdays. Not all twins returned for each measurement occasion—403 twin pairs were measured on only one occasion, 291 twin pairs returned for a second visit and 449 twin pairs were measured at all three time points (see Table 1 for complete breakdown).
Zygosity
Initially, the zygosity of twin pairs was determined by participants completing a standard questionnaire where questions about similarity were asked. In addition, genotypic zygosity diagnosis of same-sex adolescent twins was determined by typing nine independent highly polymorphic DNA microsatellite markers and the amelogenin sex marker using the Profiler multiplex marker set (AmpFLSTRR Profiler Plus T, Applied Biosystems, Foster City, CA, USA). Zygosity was assigned with a probability of error <10−4 (Nyholt 2006). Subsequently, zygosity of DZ pairs was confirmed by typing at least 400 markers genome-wide for linkage analysis.
Biometrical modelling
Genetic analysis
Classical biometrical model-fitting methods were used to decompose the total phenotypic variance in an observed trait into four sources of variance: additive genetic (A), non-additive (dominance, epistasis or recessitivity) genetic (D), environmental influences shared by members of a family (C) and environmental influences unique to each family member (E). However, C and D are confounded and thus cannot be estimated simultaneously in analyses of twins reared together, so only one of these parameters can be estimated in a model (Grayson 1989; Hewitt 1989). Since MZ twins are genetically identical, additive and non-additive genetic effects are correlated as 1.0. DZ twin pairs, like any other sibling pair, share on average half of their genes and hence are correlated as 0.5 and 0.25 for additive and non-additive genetic effects respectively. Shared environment effects correlate to an equal extent in MZ and DZ twins (i.e. correlated at 1 for members of both MZ and DZ pairs). Unique or non-shared environmental effects are by definition uncorrelated and also include measurement error.
The standard twin design can be extended to consider whether the genetic factors influencing the variation in a trait is identical among males and females, and whether there are differences in the magnitude of the same genetic and environmental variance components between the sexes. These two questions can be formally tested in a general sex-limitation model. In this model, females and males have genetic (A F, A M/D F, D M) and environmental (C F, C M/E F, E M) variables that are not constrained to be equal, allowing magnitudes of these effects to differ between the sexes. Including DZOS twins in the analyses, not only increases power, but allows one to estimate, separately, an additional male (or female, but not both) specific additive genetic, A′M (A′F), non-additive genetic, D′M (D′ F), or common environmental, C′M (C′ F) variable which does not correlate with the genetic or environmental effects on the female (or male) phenotype. Significant estimates of these sex specific effects indicate that the genes or environments which influence a trait in males are not identical to those which influence a trait in females, or vice versa. Sex-limitation models are discussed in more detail in Neale and Cardon (1992) and Medland (2004).
Longitudinal analysis
With longitudinal data it is possible to test whether a genetic factors affecting a trait is the same throughout time and/or whether “new” genes or “new” environmental factors start to operate at specific points in time. A model was fitted to the data that was able to discriminate between transient factors that affect measurement at one time point only, and factors that are continuously present or exert a long term influence throughout the time series (Boomsma et al. 1989; Neale and Cardon 1992). As such, the simplex model takes full advantage of the time series nature of longitudinal data (i.e. that causation is unidirectional throughout time; Boomsma et al. 1989) allowing testing of stronger hypotheses about development.
Every variance component structure in a simplex model is influenced by latent variables at time i, which are, in turn, influenced by the latent variable at time i - 1 (see Fig. 1). This relationship between latent variables is termed autoregressive and can be described by the equation: η i = β i η i−1 + ζ i, where η i is the latent variable at time i (i > 0), βi is the regression of the latent variable at time i on the previous latent variable at i−1, and ζ i represents a random input term (innovation) which is uncorrelated with η i−1. When using MZ and DZ twin pairs, structural equations of this type may be expressed for each latent genetic and environmental source of variation (i.e. A, E and C or D). Part of the model is a structural equation relating the observed phenotypes to the latent phenotypes: y i = λ i η i + ε i, where λ i is the loading of the observed phenotype on the latent variable at time i, and ε i is a measurement error term which affects the phenotype, but is uncorrelated with η i. In this study, the loadings of the observed variables on the latent variables (i.e. the λs) were set to unity and the variances of the innovation terms were estimated. This allows the model to be identified, whereby every parameter in the structural equation model can be estimated uniquely and consistently.

Full ADE Simplex Model for the observed variable, BMI, which is measured across three time points (at age 12, 14 and 16). The loadings of the observed variables (X) on the latent factors (λ) are set to unity and the variances of the innovations terms (ζ) are estimated. A measurement error term (ε) also influences the variance of each observed phenotype and is equated across twins and measurement occasions. The β weights are solely responsible for the covariation among variables. Correlations between co-twins for additive genetic factors are fixed to 1 for MZ twin pairs and 0.5 for DZ twin pairs. Correlations for non-additive genetic factors are 1 for MZ twin pairs and 0.25 for DZ twin pairs. Unique or non-shared environmental effects are uncorrelated between co-twins
There is an important conceptual distinction between innovations of latent variables and measurement errors of observed variables. The innovations are the part of the latent variable at time i that is not caused by the latent variable at time i - 1, but are part of every subsequent time point, i + 1, i + 2, ... i +n. In contrast, the random errors of measurement, ε, are terms that do not influence subsequent observed variables. Thus, the simplex model allows differentiation between the long-term consequence of an experience at one particular time and the continuous presence and influence of a casual factor (Neale and Cardon 1992). In addition, the variance of all measurement error terms was constrained to be equal in order for the model to be identified. This is because error variance at these occasions would otherwise be indistinguishable from innovation variance.
Variance component structures of the genetic and simplex models were estimated through structural equation modelling using the software package Mx (Neale et al. 2003). Univariate genetic models were applied to the raw data separately at each age prior to fitting the sex-limited longitudinal model. Results from these models were used as guide for the saturated sex-limited simplex model. Initially, the saturated model (ACE or ADE) was fitted to the data (i.e. maximum number of parameters). Subsequent, simplified models were fitted to test whether A, C (or D) or both parameters could be dropped from the full model. In the simplex model, the question of whether different genes or environmental influences affect the trait at different ages was investigated by removing, one at a time, the relevant innovation terms from the full model. The fit of each submodel was tested using the likelihood-ratio Chi-square test and the Akaike information criterion (AIC) (Akaike 1987), a measure of model fit relative to model parsimony. For each phenotype, the best-fitting model was identified as the model with a non-significant likelihood-ratio Chi-square probability with the largest negative AIC.
Results
Data preparation
Since maximum likelihood variance component analysis assumes that phenotypic distributions are normal, a natural logarithm transformation was performed to improve the shape of the positively skewed BMI distribution (here after called lnBMI and used in all further analyses). Family outliers (i.e. bivariate outliers) exceeding three standard deviations from the mean were identified using the Mx%P function in Mx (in which the Mahalanobis distance for each family represented as a Z-score was used) (Neale et al. 2003) at each age group (age 12, N = 5; age 14, N = 8; age 16, N = 7) and were excluded from the analysis. Interestingly, one twin pair were outliers at all three ages, a different twin pair were outliers at both 12 and 14, and another twin pair were outliers at both 14 and 16. Means and standard deviations of BMI by age group, zygosity and sex are shown in Table 2. These values are obtained after the removal of twin pair outliers.
The percentage of each age group, stratified by sex, that met the requirements for classifications as obese, overweight (according to cut-offs suggested by Cole et al. (2000)) and normal weight is shown in Table 3. Most notably, the percentage of obese adolescent boys was similar at age 12 and 14 but increased dramatically at age 16. This was also observed in adolescent girls. In addition, the age trend showed decreasing prevalence in the non-obese (or “normal”) category, with between 80 and 83% of the population falling within the “normal” BMI range.
Biometrical modelling
Preliminary analysis
Prior to model-fitting, basic assumptions concerning the equality of means and variances within twin pairs, across sex and zygosity were formally tested using Mx, in addition to hypotheses about the covariance structure. Furthermore, co-twin correlations were used to guide model selection (ACE, ADE) for biometric analyses.
Means at age 12 could be equated between twin pairs and across zygosity groups without a significant reduction in model fit. At age 14, means could be equated across same-sex zygosity groups. Means for same-sex male twins could be equated to those of the respective DZOS-twin ( \( \chi ^{2}_{2} = 2.95 \), p = 0.229), but the means for same-sex female twins could not be equated (\( \chi ^{2}_{2} = 6.52 \), p = 0.038). Means of lnBMI at age 16 could be equated across same-sex zygosity groups, but the means for same-sex male and female twins could not be equated to those of the respective DZOS twins (\( \chi ^{2}_{2} = 12.19 \), p < 0.001; \( \chi ^{2}_{2} = 16.08 \), p = 0.002, respectively) (see Table 2).
Several covariates were included in the means model: age, sex and a sex by age interaction term. Although great care was taken to measure all twins close to their 12th, 14th and 16th birthdays, there was still a small number of adolescents who were unable to be measured for height and weight at these ages due to different circumstances and, hence the inclusion of the age covariate. At all three ages, the age, sex and sex by age interaction regression coefficients could be removed from the means model without a significant loss of fit, indicating that age effects on means at all time points during puberty are the same for males and females.
At age 12 and 14, under the best-fitting means model, variances could be equated across all zygosity groups. At age 16, however, under the best-fitting means model, variances for same-sex female twins could be equated to those of the respective DZOS twin (\( \chi ^{2}_{2} = 1.63 \), p = 0.442), but variances for same-sex male twins could not be equated (\( \chi ^{2}_{2} = 8.89 \), p = 0.012). Twin–twin correlations for the best-fitting means and variance model for each age are shown in Table 4. Under the best-fitting means and variances model at all three ages, twin correlations could be equated across MZ groups and across DZ groups without reducing the fit of the model significantly. However, correlations could not be equated across zygosities at any age, indicating that genetic effects explained significant portions of the variance (\( \chi ^{2}_{1} = 243.67 \), p < 0.001 at age 12; \( \chi ^{2}_{1} = 198.81 \), p < 0.001 at age 14; \( \chi ^{2}_{1} = 203.56 \), p < 0.001 at age 16). Furthermore, familial aggregation (i.e. whether family members are more similar on the trait under investigation than unrelated individuals) was tested by setting all correlations to 0 and found to be present for lnBMI at age 12 (\( \chi ^{2}_{2} = 604.37 \), p < 0.001), 14 (\( \chi ^{2}_{2} = 470.54 \), p < 0.001) and 16 (\( \chi ^{2}_{2} = 430.99 \), p < 0.001).
Genetic analysis
After testing the basic assumptions concerning the means and variances within twin pairs, a standard univariate model was fitted to the lnBMI data for each age. The magnitude of the MZ:DZSS correlation ratios (r MZ > 2r DZSS) at age 12, 14 and 16 indicated evidence for genetic dominance (see Table 4). An ADE saturated model provided the better fit in males and females across all three ages, with variance components able to be equated across the sexes. In further simplified models, at age 12 and 14, the non-additive genetic variance could be removed from the model. However, at age 16, there was a very large non-additive genetic component, and thus the best-fitting model include additive genetic, non-additive genetic and unique environmental parameters. Variance components estimates for the saturated and best-fitting models for lnBMI are shown in Table 5. All best-fitting models displayed had the lowest AIC.
Longitudinal analysis
Male and female phenotypic correlations between the three different measurement occasions for lnBMI are shown in Table 6. Correlations for lnBMI in each sex were the highest amongst adjacent occasions and the lowest between distant occasions (i.e. between 12 and 16 years of age), consistent with a simplex model.
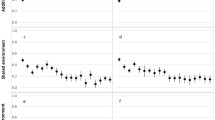
An ADE general sex-limited model was fitted to the lnBMI data, with additional female specific additive genetic (A′F) and non-additive genetic (D′F) variance components estimated separately. However, A′F and D′F could be removed from the sex-limitation model, indicating there is no significant evidence of either female specific additive genetic or female non-additive genetic influences on lnBMI (\( \chi ^{2}_{5} = 1.98 \), p = 0.852 and \( \chi ^{2}_{5} = 0.992 \), p = 0.963, respectively), and thus, an ADE simplex model provided the best fit in males and females (i.e. had the lowest AIC). Non-additive genetic and unique environmental variance components were the same across sexes (\( \chi ^{2}_{5} = 5.74 \), p = 0.332 and \( \chi ^{2}_{5} = 8.68 \), p = 0.122, respectively), whilst the additive genetic variance component differed significantly (\( \chi ^{2}_{5} = 15.58 \), p = 0.008). Simplex models for males and females are shown in Figs. 2a and 3a, respectively.

Parameter estimates (with 95% confidence intervals) for simplex models for lnBMI in males. (a) Best-fitting simplex model and (b) AE simplex model. 95% confidence intervals are given in brackets and insignificant pathways are indicated with dashed arrows. Only twin one is shown in figure. (N.B. DZOS twins are included in analysis)

Parameter estimates (with 95% confidence intervals) for simplex models for lnBMI in females. (a) Best-fitting simplex model and (b) AE simplex model. 95% confidence intervals are given in brackets and insignificant pathways are indicated with dashed arrows. Only twin one is shown in figure (N.B. DZOS twins are included in analysis)
In each variance component structure, the effects of dropping the innovation at age 14, the innovation at 16, and both innovations from the model were tested. The additive genetic structures were similar in both sexes. The most important component was the large genetic innovation influencing the first measurement occasion (i.e. at age 12). The innovations influencing the second (i.e. age 14) and third (i.e. age 16) occasions were small and could be dropped, together and separately, from the model without a significant loss in fit in males only (\( \chi ^{2}_{2} = 1.26 \), p = 0.532). In females, the additive genetic innovations at age 14 and 16 could not be removed from the model, separately or together without a significant loss in fit (\( \chi ^{2}_{2} = 12.69 \), p = 0.002).
In both sexes, there were also large transmission coefficients, indicating that most genetic variance was transmitted to subsequent ages. This in turn implies that the same genetic factors are responsible for the variation across all three measurement occasions. The non-additive genetic innovation at age 14 in males and females could be dropped from the model without deterioration in model fit (\( \chi ^{2}_{1} = 0.13 \), p = 0.721). However, innovation at age 16 could not be dropped from the model (\( \chi ^{2}_{1} = 6.20 \), p = 0.013). In addition, both innovations could not be dropped together (\( \chi ^{2}_{2} = 6.36 \), p = 0.042).
For the unique environmental structure, innovations at 14 and 16 were significant and could not be dropped without a significant loss in fit, separately or together (\( \chi ^{2}_{2} = 26.24 \), p < 0.001). The error measurements for each sex could be equated across males and females (\( \chi ^{2}_{1} = 1.65 \), p = 0.199) and could not be dropped from the model (\( \chi ^{2}_{1} = 5.10 \), p = 0.024).
In view of our small samples and consequently low power to disentangle additive and non-additive genetic variance components, we fitted an AE simplex model to males and females (Figs. 2b, 3b, respectively). Since it is known that estimates of A and D are strongly correlated in the classical twin design (r ≈ −0.91), fitting A alone will absorb most of any dominance (or additive × additive epistasis) that is present and so \({\hat A}\) (estimates of A) is a reasonable estimate of the total genetic variance. By doing so, we now see that there are significant genetic innovations at 14 and 16 (\( \chi ^{2}_{2} = 190.91 \), p < 0.001 and \( \chi ^{2}_{2} = 261.58 \), p < 0.001 for males and females, respectively), whereas in Figs. 2a and 3a these innovations were partitioned between A and D sources and neither was significant. This indicates that non-additive genetic variance has moved into the additive genetic variance component, providing evidence that using broad sense heritability to represent genetic variance (instead of both additive and non-additive genetic variance) eases interpretability when very large samples are not available.
Table 7 displays the proportion of the phenotypic variance due to innovation or transmitted from previous occasions for each source (A, D and E) of covariance. This table emphasises that most of the lnBMI phenotypic variances for males and females were due to genetic factors transmitted from previous ages.
Discussion
Our study represents one of the few longitudinal studies of twins conducted in paediatric samples to address the heritability of BMI. It adds to our currently limited knowledge of the developmental changes in genetic and environmental influences in weight regulation during puberty. We applied a general sex-limitation model that allowed for both the assessment of whether genetic effects operating longitudinally during puberty may differ between males and females, and the assessment of sex differences in magnitude of genetic and environmental variance components. Genetic analyses of adolescent longitudinal data can help distinguish the changes during development in both sexes, possibly leading to a better understanding of the genetic and environmental regulation of weight. Furthermore, longitudinal analyses increase power to detect sources of variance that are too small to be detected in univariate or cross-sectional analyses. The results from the current analyses suggest that the patterns of developmental change in gene expression in BMI are not the same in the two sexes.
One interesting finding in our study is the mean and standard deviation differences between same-sex female and same-sex male twins compared to their respective DZOS twin (see Table 2). For instance, the females of the DZOS twin pairs had a higher mean than those from the same-sex twin pairs, more noticeably at age 16. One possibility, is the likely exposure of prenatal masculinization by the hormones of the male co-twin (Resnick et al. 1993; Miller 1994; Loehlin and Martin 1998, 2000). Evidence from numerous studies have found that for several litter-bearing mammals, female foetuses exposed prenatally to male hormones, when located between two male foetuses, may show masculinized effects on a variety of anatomical and behavioural characteristics, compared to female foetuses that have been located between two female foetuses (vom Saal 1989). However, evidence of this occurring in humans across different traits, particularly in personality traits, has been inconclusive (Burns et al. 1992; McFadden 1993; Resnick et al. 1993; Miller 1994; Rodgers et al. 1998).
In this study, a longitudinal model comprising additive genetic (A), non-additive genetic (D) and unique environmental (E) variances provided the best fit to the adolescent male and female data. Sex differences were found in the magnitude of additive genetic variances between males and females, but the non-additive genetic and unique environmental variances were equal across sexes. Furthermore, no female-specific genetic or environmental effects were found. Most of the variation in BMI at all three ages was explained by genetic (additive and/or non-additive) influences for males and females, confirming findings from other twin studies (Loesch et al. 1995; Huggins et al. 2000). In males, the only input of additive genetic influence for BMI occurs at age 12 and consequently this genetic variance influenced BMI at subsequent ages. However, in females, as well as a substantial influence at age 12, new sources of additive genetic variances were present at age 14 and 16.
Epidemiologic observations of the relative constancy of body composition over long periods of time support strongly a biological basis for the regulation of body fat. The additive genetic variance observed in the sex-limitation models may be explained by several regulatory and structural genes responsible for weight regulation during puberty. Additionally, differences were found between males and females in the timing of largest amount of weight gain (largest mean change) and in the period of maximum genetic influence on BMI. These differences are consistent with known sex differences in the role of leptin in regulation of food intake, weight gain and body fat distribution. Leptin is known to control body fat by co-ordinated regulation of feeding behaviour, metabolic rate, autonomic nervous system regulation and body energy balance. Past research has found that there are important age and gender-based differences in the regulation and action of leptin during childhood and adolescence (Ong et al. 1999; Apter 2003).
Throughout puberty, non-additive genetic variance could be equated across males and females, suggesting that significant epistatic, dominant or recessive effects influenced BMI similarly in the sexes at 12, 14 and 16. However, in the univariate models at age 12 and 14, non-additive genetic influences could be removed as a source of variation for male and female BMI. This could be the result of type II errors due to reduced power in univariate models compared to longitudinal models (Martin et al. 1978). Most interestingly, during development, the percentage of non-additive genetic effects influencing BMI increased with age (see Table 7). Similar results were found in a sample of young Polish twins, where there was an increasing non-additive genetic component, especially after age 16 (Huggins et al. 2000). In a young Australian twin sample (aged 18–30), genetic dominance (or other genetic non-additivity) explained most of the variance of BMI in males and females (Neale and Cardon 1992). The interaction of sex steroids, hormones, leptin production, insulin and growth hormones may explain the non-additive genetic influences observed in the current study. Furthermore, non-additive genetic variance may represent gene–gene interaction occurring during puberty when many metabolic roles of hormones are controlled by a complex interaction of genes which cause a direct or indirect influence on lipid metabolism, the amount of adiposity and its regional distribution (Roemmich and Rogol 1999).
Common environmental influences were not important in this study in the variation in BMI in males and females during development. But unique environment accounted for a small percentage of the overall phenotypic variation in BMI. Other research has also shown that unique environmental effects have a considerable influence on obesity, whereas common environmental influences are small or negligible (Jacobson and Rowe 1998). This does not rule out the possibility that there are common environmental influences on BMI during puberty, but the effects must be small relative to non-additive genetic effects (Grayson 1989; Hewitt 1989). In western societies, numerous environmental influences may play a part in weight regulation of young adults. Unique or non-shared environmental influences might include dietary habits such as severe dieting and eating disorders, fitness routines and sporting exercise, interactions with siblings and the influence of television and other media, in addition to stochastic epigenetic influences. The results from this study suggest that the unique environmental sources were the same in males and females. Most interestingly, the percentage of environmental variation transmitted from previous ages to age 16 was the same, suggesting that unique habits occurring at age 12 and 14 continue to have an effect later in puberty. Booth et al. (1998) noted that patterns of physical activity learned during adolescence are generally carried into adulthood. Thus, activities that promote sedentary behaviours should be kept to a minimum to prevent unfavourable weight gain later in life.
Limitations and future directions
Whilst reliable pubertal status was available in girls (age at menarche were collected), no reliable information was obtained for boys (presence of auxiliary hair was used as opposed to age at spermache), and hence, this information was not used in the current analyses. Furthermore, in preliminary analyses, the current accessible information was not found to be significant within the means model. However, in a study by Anderson et al. (in press) the estimation of the average age at menarche for the majority of the girls included in this sample was 154.9 months (approximately 12.91 years), although it is unclear what is the average age at spermache due to lack of data. Nevertheless, once reliable pubertal status becomes available for boys, further investigation of these analyses is warranted to provide perhaps more accurate results.
The main limitation in this study is the lack of power, despite our relatively large sample size of over 1,100 twin pairs. Our results highlight that very large samples are needed to discriminate between additive and non-additive genetic parameters in twins; over 1,000 twin pairs are needed to detect non-additive genetic variance that accounts for at least 20% of the total variation (Martin et al. 1978). However, in the current case, we had less than 1,000 pairs in each univariate model, and in addition, fewer than 1,000 pairs were measured at all three ages in the longitudinal model (see Table 1). With respect to power, additive genetic variance and non-additive genetic variance are negatively confounded. That is, if one of these variance components is removed, then the variation explained by that parameter is expected to move to the other source of genetic variation. Therefore the heritabilities reported should be interpreted as broad sense genetic heritabilities encompassing the sum of additive and non-additive genetic factors, giving a true estimate of overall genetic variance. When taken in this broad sense, there are now appreciable genetic innovations (AI + DI; see Table 7, Figs. 2b, 3b) at age 14 and 16, that would not have been significant if the genetic source had been separated into additive and non-additive genetic variance.
Results from this study suggest that genes play an important part in the variation in BMI during puberty. Our analysis also suggests that the genetic architecture of BMI may indeed be different across sexes, perhaps resulting from selective processes during human evolution. Covariation between the measures across time in males and females was mostly due to the same genetic factor, making it possible to use multivariate methods to identify likely QTLs. Identification of these genes, and understanding their physiological pathways and potential interactions with environmental factors, may inform appropriate prevention strategies for childhood and adolescent obesity, and contribute to a better understanding of how and why obesity develops later in life.
References
Akaike H (1987) Factor analysis and AIC. Psychometrika 52:317–332
Allison DB, Heshka S, Neale MC, Heymsfield SB (1994a) Race effects in the genetics of adolescents’ body mass index. Int J Obes Relat Metab Disord 18(6):363–368
Allison DB, Heshka S, Neale MC, Lykken DT, Heymsfield SB (1994b) A genetic analysis of relative weight among 4,020 twin pairs, with an emphasis on sex effects. Health Psychol 13(4):362–365
Anderson CA, Duffy DL, Martin NG, Visscher PM (in press) Estimation of variance components for age at menarche in twin families. Behav Genet
Apter D (2003) The role of leptin in female adolescence. Ann N Y Acad Sci 997:64–76
Australian Institute of Health and Welfare (AIHW) (2000) Australia’s health. Canberra
Bodurtha JN, Mosteller M, Hewitt JK, Nance WE, Eaves LJ, Moskowitz WB, Katz S, Schieken RM (1990) Genetic analysis of anthropometric measures in 11-year-old twins: the Medical College of Virginia Twin Study. Pediatr Res 28(1):1–4
Boomsma DI, Martin NG, Molenaar PC (1989) Factor and simplex models for repeated measures: application to two psychomotor measures of alcohol sensitivity in twins. Behav Genet 19(1):79–96
Booth ML, Macaskill P, Phongsavan P, McLellan L, Okely T (1998) Methods of the NSW schools fitness and physical activity survey, 1997. J Sci Med Sport 1(2):111–124
Borecki IB, Higgins M, Schreiner PJ, Arnett DK, Mayer-Davis E, Hunt SC, Province MA (1998) Evidence for multiple determinants of the body mass index: the National Heart, Lung, and Blood Institute Family Heart Study. Obes Res 6(2):107–114
Bouchard C, Savard R, Despres JP, Tremblay A, Leblanc C (1985) Body composition in adopted and biological siblings. Hum Biol 57(1):61–75
Burns EM, Arehart KH, Campbell SL (1992) Prevalence of spontaneous otoacoustic emissions in neonates. J Acoust Soc Am 91(3):1571–1575
Chambers ML (2001) Height, weight and body mass index. In: Emde RN, Hewitt JK (eds) Infancy to childhood: genetic and environmental influences and developmental change. Oxford University Press, New York
Cole TJ, Bellizzi MC, Flegal KM, Dietz WH (2000) Establishing a standard definition for child overweight and obesity worldwide: international survey. BMJ 320(7244):1240–1243
Cornes BK, Medland SE, Ferreira MA, Morley KI, Duffy DL, Heijmans BT, Montgomery GW, Martin NG (2005) Sex-limited genome-wide linkage scan for body mass index in an unselected sample of 933 Australian twin families. Twin Res Hum Genet 8(6):616–632
Deng HW, Deng H, Liu YJ, Liu YZ, Xu FH, Shen H, Conway T, Li JL, Huang QY, Davies KM, Recker RR (2002) A genomewide linkage scan for quantitative-trait loci for obesity phenotypes. Am J Hum Genet 70(5):1138–1151
Eckersley RM (2003) Losing the battle of the bulge: causes and consequences of obesity. Healthlink Health Promot J ACT Reg 1–3
Grayson DA (1989) Twins reared together: minimizing shared environmental effects. Behav Genet 19(4):593–604
Harris JR, Tambs K, Magnus P (1995) Sex-specific effects for body mass index in the new Norwegian twin panel. Genet Epidemiol 12(3):251–265
Hewitt JK (1989) Of biases and more in the study of twins reared together: a reply to grayson. Behav Genet 19(4):605–608
Hill JO, Wyatt HR, Reed GW, Peters JC (2003) Obesity and the environment: where do we go from here? Science 299:853–855
Huggins RM, Hoang NH, Loesch DZ (2000) Analysis of longitudinal data from twins. Genet epidemiol 19(4):345–353
Jacobson KC, Rowe DC (1998) Genetic and shared environmental influences on adolescent BMI: interactions with race and sex. Behav Genet 28(4):265–278
Korkeila M, Kaprio J, Rissanen A, Koskenvuo M (1991) Effects of gender and age on the heritability of body mass index. Int J Obes 15(10):647–654
Loehlin JC, Martin NG (1998) A comparison of adult female twins from opposite-sex and same-sex pairs on variables related to reproduction. Behav Genet 28(1):21–27
Loehlin JC, Martin NG (2000) Dimensions of psychological masculinity-femininity in adult twins from opposite-sex and same-sex pairs. Behav Genet 30(1):19–28
Loesch DZ, Hopper JL, Rogucka E, Huggins RM (1995) Timing and genetic rapport between growth in skeletal maturity and height around puberty: similarities and differences between girls and boys. Am J Hum Genet 56(3):753–759
Maes HH, Neale MC, Eaves LJ (1997) Genetic and environmental factors in relative body weight and human adiposity. Behav Genet 27(4):325–351
Martin NG, Eaves LJ, Kearsey MJ, Davies P (1978) The power of the classical twin study. Heredity 40(1):97–116
McFadden D (1993) A masculinizing effect on the auditory systems of human females having male co-twins. Proc Natl Acad Sci USA 90(24):11900–11904
Medland SE (2004) Alternate parameterization for scalar and non-scalar sex-limitation models in Mx. Twin Res 7(3):299–305
Meyer JM (1995) Genetic studies of obesity across the life span. In: Turner RJ, Cardon LR, Hewitt JK (eds) Behaviour genetic approaches in behavioural medicine. Plenum Press, New York, pp 145–167
Miller EM (1994) Prenatal sex hormone transfer: a reason to study opposite-sex twins. Pers Individ Dif 17:511–529
Neale MC, Cardon LR (1992) Methodology for genetic studies of twins and families. Kluwer Academic Publishers, Dordrecht
Neale MC, Boker SM, Xie G, Maes HH (2003) Mx: statistical modeling, 6th edn. Department of Psychiatry, Virginia Commonwealth University, Richmond
Nyholt DR (2006) On the probability of dizygotic twins being concordant for two alleles at multiple polymorphic loci. Twin Res Hum Genet 9(2):194–197
Ong KK, Ahmed ML, Dunger DB (1999) The role of leptin in human growth and puberty. Acta Paediatr Suppl 88(433):95–98
Perola M, Ohman M, Hiekkalinna T, Leppavuori J, Pajukanta P, Wessman M, Koskenvuo M, Palotie A, Lange K, Kaprio J, Peltonen L (2001) Quantitative-trait-locus analysis of body-mass index and of stature, by combined analysis of genome scans of five Finnish study groups. Am J Hum Genet 69(1):117–123
Pietilainen KH, Kaprio J, Rissanen A, Winter T, Rimpela A, Viken RJ, Rose RJ (1999) Distribution and heritability of BMI in Finnish adolescents aged 16y and 17y: a study of 4884 twins and 2509 singletons. Int J Obes Relat Metab Disord 23(2):107–115
Resnick SM, Gottesman II, McGue M (1993) Sensation seeking in opposite-sex twins: an effect of prenatal hormones? Behav Genet 23(4):323–329
Rodgers CS, Fagot BI, Winebarger A (1998) Gender-typed toy play in dizygotic twin pairs. Sex Roles 39:173–184
Roemmich JN, Rogol AD (1999) Hormonal changes during puberty and their relationship to fat distribution. Am J Hum Biol 11(2):209–224
Schousboe K, Willemsen G, Kyvik KO, Mortensen J, Boomsma DI, Cornes BK, Davis CJ, Fagnani C, Hjelmborg J, Kaprio J, de Lang M, Luciano M, Martin NG, Pedersen N, Pietilainen KH, Rissanen A, Saarni S, Sorensen TIA, van Baal CM, Harris JR (2003) Sex differences in heritability of BMI: a comparative study of results from twin studies in eight countries. Twin Res 6(5):409–421
Stunkard AJ, Harris JR, Pedersen NL, McClearn GE (1990) The body-mass index of twins who have been reared apart. N Engl J Med 322(21):1483–1487
vom Saal FS (1989) Sexual differentiation in litter-bearing mammals: influence of sex of adjacent fetuses in utero. J Anim Sci 67(7):1824–1840
Wright M, Geus ED, Ando J, Luciano M, Posthuma D, Ono Y, Hansell N, Van Baal C, Hiraishi K, Hasegawa T, Smith G, Geffen G, Geffen L, Kanba S, Miyake A, Martin N, Boomsma D (2001) Genetics of cognition: outline of a collaborative twin study. Twin Res 4:48–56
Zhu G, David DL, Eldridge A, Grace M, Mayne C, O’Gorman L, Aitken JF, Neale MC, Hayward NK, Green AC, Martin NG (1999) A major quantitative-trait locus for mole destiny is linked to the familial melanoma gene CDKN2A: a maximum-likelihood combined linkage and association analysis in twins and their sibs. Am J Hum Genet 65:483–492
Acknowledgements
The author would like to acknowledge the help and support provided by many individuals at the Queensland Institute of Medical Research including: Ann Eldridge and Marlene Grace for phenotype collection, Alison MacKenzie for project coordination, David Smyth for data management and the twins for their generous participation. The Twin Mole study was funded by grants from the Queensland Cancer Fund, National Health and Medical Research Council (901061 and 950998) and the Cooperative Research Centre for Discovery of Genes for Common Human Diseases. B.K.C. is supported by an NHMRC Public Health Scholarship (361302).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by David Allison.
Rights and permissions
About this article
Cite this article
Cornes, B.K., Zhu, G. & Martin, N.G. Sex Differences in Genetic Variation in Weight: a Longitudinal Study of Body Mass Index in Adolescent Twins. Behav Genet 37, 648–660 (2007). https://doi.org/10.1007/s10519-007-9165-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10519-007-9165-0