
Abstract
Oral squamous cell carcinoma (OSCC) is a malignant tumor with high mortality and poor prognosis. Many OSCC patients have low response rate to current treatments including immunotherapies largely due to the immune-suppressive tumor microenvironment (TME). Chemotherapy could induce immunogenic cell death (ICD), a type of cell death such as pyroptosis and necroptosis, which has proved to be capable to alter the immune-suppressive TME and beneficial for better anti-tumor effect. GSDME, a key protein of pyroptosis, is however often silenced in tumors due to abnormal methylation. To overcome these limitations, we utilizied methyltransferase inhibitor (decitabine, DAC) to trigger pyroptosis of tumor cells, combined with chemodrug cisplatin (DDP) and immune checkpoints inhibitors to amplify the immunotherapies outcomes. To the best of our knowledge, this is the first study of tumor suppressive effect of GSDME in OSCC. Our investigation demonstrated that stimulation of GSDME expression could improve the sensitivity of chemotherapeutics, activate inflammatory tumor cell pyroptosis and alter the tumor immune-suppressive microenvironment, providing an important perspective for clinical OSCC treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Head and neck cancer is one of the most common types of cancer worldwide. Most of these cancers are squamous cell carcinomas (SCC) that arise from epithelial cells on the mucosal surfaces of the oral cavity, larynx, oropharynx, and hypopharynx [1]. Oral squamous cell carcinoma (OSCC) is the predominant subtype of head and neck squamous cell carcinoma (HNSCC), with approximately two-thirds of cases occurring worldwide. OSCC is usually diagnosed at an advanced stage with high mortality and poor prognosis. Currently, the common treatments include surgical and chemoradiotherapy. However, many patients after surgery and chemotherapy still experienced cancer recurrence as well as distant metastasis [2]. Current standard first-line treatments for OSCC, including systemic chemotherapy combined with cisplatin (DDP), carboplatin, 5-fluorouracil (5-FU) and cetuximab could provide a median overall survival of approximately 10 months [3]. It is urgently needed to develop more effective treatments for OSCC patients.
Recent studies demonstrated that tumor microenvironment (TME) has become an important factor affecting tumor progression and clinical recurrence in OSCC patients [2]. A better understanding of the interaction between OSCC TME, especially cancer cells and their surrounding TME, will provide precise treatment for patients. Solid malignancies include not only tumor cells, but also surrounding cells such as vascular endothelial cells, fibroblasts, a variety of immune cells and extracellular soluble molecules (cytokines, chemokines, growth factors). Most of immune cells in TME are immunosuppressive cells, such as tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), regulatory T cells (Treg) and fewer anti-tumor effector cells, such as cytotoxic CD8+ T cells and natural killer cells (NK), etc. [4]. Recently, TME has been divided into “cold and hot”. “Cold” refers to non-inflammatory TME and “hot” refers to inflammatory TME, which is mainly attributed to the production of pro-inflammatory cytokines and T cells infiltration. “Hot” tumors are characterized by sufficient T cells infiltration and immune activation, while “cold” tumors show significant features of T cells deficiency [5]. OSCC also has pathological features of “cold” tumors, less effector lymphocyte infiltration and immunosuppression [6].
Turning the immunosuppression TME has proved to achieve improved anti-tumor efficiency. Current clinical treatment for recurrent and metastatic OSCC usually included combination standard therapy (cisplatin-based chemoradiotherapy) and immunotherapy (anti-PD-1, anti-PD-L1, anti-CTLA-4, anti-LAG-3, etc.) [7, 8]. Such combination therapies are able to activate functional immune cells, such as T cells, DC cells and NK cells, etc., meanwhile reduce the infiltration of immunosuppressive cells, turn “cold” tumors into “hot” tumors. However, simply application of immunocheckpoint blockades only achieve moderate therapeutic efficacy, more effective strategies to alter the immunosuppressive OSCC TME are still needed.
Immunogenic cell death (ICD) plays an important role in host protection against pathogens and maintenance of homeostasis. Unlike apoptosis, ICD is a immunocompetent type of cell death, characterized by damage or rupture of cell membranes and release of inflammatory to stimulate the surrounding environment [9]. Pyroptosis is a type of ICD that could be used as a host defense against pathogenic infections, leading to the release of pro-inflammatory cytokines through membrane pores formed by gasdermin family members mediating the formation of inflammasomes and the activation of inflammatory asparaginase [10]. Gasdermin E (GSDME) is a member of the gasdermin family of pyroptosis execution proteins, originally thought to be associated with autosomal dominant inheritance of inherited hearing loss [11]. Methylation of the promoter region of GSDME DNA leads to its silencing in many tumor cells. Small molecule inhibitors [12] or chemotherapy drugs [13], promoted caspase3 activated to induced cleaved of GSDME, GSDME directly transformed non-inflammatory cell apoptosis into pyroptosis through the formation of pore permeable plasma membrane. Generally, apoptotic cells were eliminated by neighboring phagocytes due to loss of membrane integrity and are considered as non-immunogenic cells, while pyroptosis can mediate membrane rupture and exudation of contents causing an inflammatory response. Meanwhile, it has also been documented that overexpression of GSDME can recruit more immune cells to reach the tumor site in OSCC[14]. This suggested that GSDME can play an important role as a tumor suppressor by changing tumor microenvironment. In fact, the presence of GSDME in tumors has been shown to increase the recruitment of immune cells [14, 15]. Therefore, activation of GSDME has important clinical implications by transforming "non-inflammatory" or "cold" tumors into "hot" tumors, thereby enhancing immunotherapy response.
In the present work, we used GSDME-deficient mice in an orthotopic OSCC mouse model to determine the immune enhancing effects of GSDME. In addition, we also focused on the application of methylation inhibitors to burst GSDME expression and trigger pyroptosis of OSCC in the chemotherapy. Finally, the therapeutic efficacy of GSDME induced pyroptosis was investigated in vivo. Our study here provided a more reliable reference for subsequent clinical treatment of OSCC.
Materials and methods
Cell lines culture and treatment
The OSCC cell lines (HN6 and cal27) were stored in the central Laboratory of Peking University Stomatology Hospital. Cells were cultured in DMEM-F12 with 10% FBS (GIBCO, Life Technologies), 100 U/mL penicillin (Invitrogen) and 100 U/mL streptomycin (Invitrogen) at 37 °C with 5% CO2. To investigate the pyroptosis level induced by standard chemodrugs, HN6 and cal27 cells were treated with cisplatin (cat# 479306, Sigma Aldrich). For the intervention experiment, cells were preincubated with 5 or 10 μM Decitabine (cat# S1200, Selleck) for 24 h, 3 or 6 days. For inhibition of cell pyroptosis, cells were preincubated with 20 μM Z-DEVD-FMK (special Caspase3 inhibitor, cat# S7312, Selleck) for 1 h before treatment with above mentioned drugs.
Mice
GSDME-deficient (GSDME−/−, KO) C57BL/6 mice were grifted from Professor Shao Feng’group, Beijing Institute of Life Sciences. Female C57BL/6 mice (4–6 week) were from Beijing Vital River Laboratory Animal Technology Co., LtD (Beijing, China) and housed in the animal facility of Peking University School of Stomatology under pathogen-free conditions. The experiments were carried out under controlled conditions with a 12 h light/dark cycle. All animal experiments were performed in accordance with protocols approved by the Medical Ethics Committee and Biosafety Management Committee of Peking University (approval number LA2019197).
OSCC mouse model
To build the orthotopic OSCC tumor model, 4–6 weeks female mice were treated drinking water with 4-NQO (4-nitroquinoline N-oxide, cat# N8141, 100 μg/mL, Sigma Aldrich) for 16 weeks and then given normal drinking water for 8 weeks.
In vivo anti-tumor therapy
All OSCC model mice were randomly divided into 6 groups (n = 6). These mice were injected intravenously with PBS, DDP, anti-PD-L1 (cat# 10F.9G2, Bio X Cell), DAC + DDP, DDP + anti-PD-L1, DAC + DDP + anti-PD-L1. The dose was DAC, 10 mg/kg; DDP, 5 mg/kg; anti-PD-L1, 1 mg/kg. DAC was administered for 3 consecutive days followed by DDP/anti-PD-L1 for 2 days afterwards, total for 4 weeks. The tumor volume was defined as: V = ((length) × (width)2)/2. After treatment, the mice were sacrificed. Peripheral blood, tumor tissues and cervical lymph nodes were collected for further experiments.
Western blotting (WB)
Cells were collected and suspended in RIPA lysis buffer (cat# R0010, Solarbio) and stored at − 80 °C. The total protein concentration was measured by BCA protein assay kit (cat# 23227, Thermo Scientific). Samples were denatured in loading buffer (cat# 8015011, DAKEWE). Samples were separated with SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membrane and blocked. The PVDF membranes were incubated overnight with indicated antibodies (GSDME (cat# ab215191, Abcam), caspase3 (cat# 14220, CST) and β-actin (cat# AC038, Abclonal)) at 4 °C and then incubated with a secondary antibody at room temperature for 1 h. Immunostaining was developed by using the ECL detection system (cat# 8061011, DAKEWE). The density of protein bands was quantified and analyzed by Image Lab software.
Apoptosis assay
OSCC cell lines were treated with different drugs, incubate with Annexin-V (cat# 640,914, BioLegend) for 15 min in the dark at 4 °C, propidium iodide (PI) was added, followed by flow cytometry analysis.
TUNEL (TdT-mediated dUTP Nick-End Labeling) assay
All procedures were performed according to the TUNEL Apoptosis Detection Kit (Alexa Fluor 488) (cat# 40307ES20, TEASEN). Images of TUNEL positive signals were obtained by laser scanning confocal microscopy (Nikon A1R).
LDH release assay
LDH release assays were performed according to LDH Cytotoxicity Assay Kit (cat# 40209ES76, YEASEN). Cells were cultured in 96-well plate in advance and treated according to the different experimental plan. Reaching the predetermined time, centrifuge the cell culture plate at 400 g for 5 min, take out 120 μL from each well to a new 96-well plate and detected the sample. The absorbance was measured at 490 nm. Each sample is tested for three times and take the mean value.
ELISA (enzyme-linked immunosorbent assay) assay
ELISA assays were performed according to IL-1β Assay Kit (cat# MM-0181H2). Cells were treated with drugs and collected cell supernatants, centrifuged to remove debris and suspended cells, and then tested. Results were detected at the absorbance of 450 nm.
Immunohistochemistry and immunofluorescence
Tumor tissues were obtained from experimental mice, which were fixed with 4% paraformaldehyde and embedded in paraffin. Paraffin-embedded samples were sectioned (4 μm) and fixed on glass slides. All procedures were performed according to the Hematoxylin–Eosin (HE) Staining Kit (cat# G1120, Solarbio).
HN6 and cal27 were inoculated into 12-well plates using cell slides and treated with drugs. After treatment, cells were fixed with paraformaldehyde at room temperature for 30 min, washed with PBS three times and permeabilized with 0.1% Trition, followed by blocking solution at 4° for 1 h. After the end of blocking, the primary antibody was incubated overnight at 4 °C, and the secondary antibody was incubated the next day, followed by photography and observation (Nikon).
Multiplexed immunohistochemistry (mIHC)
All procedures were performed according to the Multiple Fluorescent Immunohistochemistry Kit (Panovue). For mIHC, a multiplex tyrosine signal amplification method. Sections were deparaffinized with xylene, graded ethanol series for 10 min and washed with TBST for 5 min, three times. To retrieve and expose antigens, a full power microwave was applied to the citrate-buffered solution for 2 min and low power for 15 min, followed by washing. Next, sections were blocked with goat serum, incubated with primary antibody (CD11c, cat# 45581T; cleaved-caspase3, cat# 9664T; CD8a, cat# 98941T, CST) overnight at 4 °C. Next day, secondary polymer horseradish peroxidase-conjugated anti-primary antibody species antibody was incubated for 1 h at room temperature and then washed. Finally, the fluorescent dye signal amplification reagent was used for 30 min incubation, covalent bond stabilization, and washing. The antigen extraction and blocking steps were repeated, followed by staining of each additional marker. Single-stained slides were included in each antibody as controls.
Isolation and culture of mouse dendritic cells
WT/KO mice were sacrificed by cervical dislocation, soaked in 75% alcohol for 5 min, the femurs and tibias of mice were removed under aseptic conditions. Both ends of long bones were cut off, the bone marrow cavity was repeatedly washed with PBS drawn with a syringe until the bones turned white, the washed cells were collected. The bone tissue was removed with a 70 μm cell strainer, the filtrate cell was centrifuged and lysed with 2 mL red blood cell lysate (cat# 420301, Biolegend) for 5 min, washed with PBS twice after centrifugation, the cells were resuspended with RPMI-1640 complete medium, counted and seeded in a 24-well plate. GM-CSF (20 ng/mL, cat# 315-03, PeproTech) and IL-4 (10 ng/mL, cat# 214-14, PeproTech) were placed in an incubator for 6 days and half change medium every two days. Results tested by flow cytometry.
T cell isolation and sorting
Tumor infiltrating lymphocytes (TIL) were harvested from mouse tongue tissue using a mouse tumor dissociation kit (cat# LTS1092P, TBD sciences) according to the manufacturer's protocol. Lymphocytes from cervical lymph nodes of mice were obtained by tissue grinding and resuspended into single cells with RPMI-1640 complete medium. The separation of mouse peripheral blood lymphocytes was performed according to the instructions for use of mouse lymphocyte separation solution (cat# 7211011, DAKEWE). CD8+ T cells from MBMCs were isolated by AriaIII (BD) with mouse CD8 antibody (cat# 100707, Biolegend). CD8+ T cells were activated with anti-CD3/CD28 antibody (cat# 100339, cat# 102115, Biolegend), cultured with 100 IU/mL IL-2 (cat# C047, Novoprotein).
Flow cytometry and intracellular cytokine staining
Immune cells were stained with fluorescence-labeled antibodies (CD45, cat# 103133; CD3, cat# 100235, cat# 100203; CD4, cat# 100509; CD8, cat# 100707; cat# 100733; CD19, cat# 152407; CD25, cat# 102033; FOXP3, cat# 320007; IL-17A, cat# 506915; F4/80, cat# 123109; CD11b, cat# 101205; Gr-1, cat# 108411; CD11c, cat# 117309; CD80, cat# 104705; CD86, cat# 105007; IL-2, cat# 503825; IFN-γ, cat# 505807; TNF-α, cat# 506307; Granzyme B, cat# 372213; CD44, cat# 103031; CD62L, cat# 104417; NK1.1, cat# 108707) all purchased from Biolegend company. Cells were collected by centrifugation and washed by staining buffer twice. Fluorescent antibodies were used to stain at 4 °C in the dark for 20 min. To analyze intracellular cytokines, cells were stimulated with Cell Activation Cocktail (with Brefeldin A, cat# 423303, Biolegend) for 4–6 h. Surface antibodies were stained for 20 min and 4% formalin was added. After washing with permeabilization washing buffer (cat# 421002, Biolegend), cells were stained with endogenous factor antibody for 20 min. Data were obtained on LSRFortessa (BD Biosciences) or cytoflex flow cytometry (Beckman Coulter). Data analyzed by FlowJo software (version 10.6.2) and Cytobank (https://premium.cytobank.cn).
Real-time quantitative polymerase chain reaction (q-PCR)
Total cellular RNA was extracted using TRIzol (cat# 15596026, Invitrogen). RNA quality and concentration were detected using NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). For amplification, 1 μg of total RNA was reverse transcribed into cDNA using the Primescript RT kit (cat# 037a, TakaRa). Real-time PCR (qRT-PCR) was performed using a 7500 PCR system (Thermofisher) followed by quantitative analysis of the data by 2−∆∆Ct. Primer sequences used were listed in Table S1.
Plasmid transfection
According to the manufacturer's instructions, empty vector (EV) and target GSDME shRNA and overexpression plasmids (OE) were transfected into OSCC cell lines using lipofectamine 3000 (Invitrogen). In order to stabilize expression, puromycin was used to screen for stable expression. These structures were synthesized by PPL (Public Protein/Plasmid Library, China).
Statistics
Data were presented as mean ± standard deviation and were performed at least in triplicate for each condition in three independent assays. Data analysis was performed using Student's t-test or one-way analysis of variance (ANOVA), unless specified. The differences were considered be statistically significant for a P < 0.05. All data were analyzed using GraphPad Prism 9 software (GraphPad, Inc). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Results
DDP induced pyroptosis in OSCC cell lines through caspase3/GSDME pathway
Pyroptosis has been reported in many tumors, such as gastric cancer [16], colon cancer [17] and melanoma [15], etc. Pyroptosis is featured by cell membrane bubble expansion until rupture [10]. DDP is a commonly used first-line drug in the clinical treatment of OSCC [18]. To verify pyroptosis in OSCC during DDP treatment, HN6 and cal27 cells were treated with DDP (20 μM) for 24 h. The tumor cells appeared bubbling after DDP treatment through light microscopy images, indicating pyroptosis in these cells (Fig. 1a). Immunofluorescence showed that GSDME protein was distributed in the cytoplasm of HN6 and cal27 cells before DDP treatment and appeared scattered punctate after DDP treatment, indicating lysis of GSDME in cytoplasm (Fig. 1b). During pyroptosis, propidium iodide (PI) could enter cells to stain DNA due to increased permeability of cell membrane and phosphatidylserine (PS) on the inner side of cell membrane could be stained by Annexin V. Therefore, Annexin V and PI double-positive consider to be pyroptosis [13]. To further validate the cascade relationship between caspase3 and GSDME cleavage, HN6 and cal27 cells were pretreated with the Z-DEVD-FMK. In addition, empty vector (EV) plasmids, overexpression (OE) plasmids and shRNA plasmids targeting GSDME were constructed in cal27 and HN6 cells. These cells were stably selected with puromycin and transfection efficiency was tested by WB assay (Supplementary Fig. 1). Flow cytometry results showed that DDP induced pyroptosis decreased after Z-DEVD-FMK treatment or si-GSDME treatment (Fig. 1c, d). LDH release were also significantly inhibited (Fig. 1e). GSDME cleavage was alleviated after pretreatment with Z-DEVD-FMK (Fig. 1f). In summary, these results demonstrated moderate pyroptosis in OSCC after DDP treatment through caspase3/GSDME pathway.

Cisplatin induce pyroptosis in OSCC cell lines through caspase3/GSDME pathway. A Light microscopy showing the pyroptosis of OSCC cell lines after DDP treatment, cell membrane burst and bubbles appeared, arrows show bubbles emerging from the plasma membrane (scale bar = 50 μm); B Location and cleavage of GSDME protein before and after DDP treatment were observed by immunofluorescence (scale bar = 10 μm); C, D Flow cytometry detected the proportion of pyroptosis in OSCC, Annexin V and PI double positive indicate pyroptosis; E LDH assay after different treatment; F Cleavaged of GSDME and caspase3 by DDP in HN6 and cal27 cells by WB assay
Inducing GSDME expression in OSCC increase the sensitivity and pyroptosis of chemotherapy
Dysregulation of epigenetic modification was a hallmark of cancer, particularly DNA methylation [19]. Many tumor cells expressed lower GSDME than normal cells, which was attributed to the highly aberrant methylation of the GSDME [20, 21]. DNA methyltransferase (DNMT) inhibitors, such as decitabine (DAC), had clinical benefits in both hematologic malignancies and solid tumors, particularly myelodysplastic syndrome (MDS) [22] and acute myeloid leukemia (AML) [23].
To validate the demethylation of GSDME by DAC, HN6 and cal27 cells were pretreated with DAC for 3 days and the level of GSDME expression was assessed by q-PCR (Fig. 2a) and WB assays (Fig. 2b, c, Supplementary Fig. 2, 3). Compared with untreated cell lines, the expression of GSDME was significantly enhanced in tumor cells after DAC pretreatment. The expression of GSDME in DAC treated cells also followed a dose dependent manner. The expression of GSDME was highest after 3 days of 10 μM pretreatment, therefore, this concentration was applied in later studies.

Inducing GSDME expression in OSCC could increase the sensitivity and pyroptosis of chemotherapy. A q-PCR detected the expression of GSDME after DAC treatment (10 μM DAC for 24 h, 3 day and 6 day); B, C WB detected the expression of GSDME after DAC treatment (10 μM DAC for 24 h, 3 day and 6 day) and statistical result in cal27; D, E Flow cytometry detected the proportion of pyroptosis in cal27 with different treatment groups (ctrl, DDP, OE-GSDME + DDP, DAC, DAC + DDP, sh-GSDME + DDP)
Flow cytometry results showed that the pyroptosis level of OE-GSDME treated with low concentration of DDP was about 70% in cal27 cells (10 μM, half of the previous experimental concentration, but the proportion of pyroptosis was twice than before), indicating that high expression of GSDME could enhance the sensitivity of OSCC cells to chemodrugs and amplify pyroptosis. In addition, DAC pretreated tumor cells after same concentration of DDP treatment, pyroptosis ratio reached to 50%. While cells treated with sh-GSDME, the proportion of pyroptosis decreased to 15% (Fig. 2d, e; Supplementary Fig. 4). Taken together, these results indicated that DAC enhanced the sensitivity of tumor cells to chemodrugs and increase pyroptosis through amplification of GSDME expression.
Combination of DAC with chemodrugs demonstrated enhanced antitumor immunity and improved antitumor effect
Immunotherapy, especially immune checkpoint blockades (ICB), such as PD-1 / PD-L1 antibodies, has been approved for more than 20 cancer types [24] including OSCC [25], which significantly changed the therapeutic prospects for OSCC [26]. However, low response rate was a major limitation of ICB treatment, which hindered the application of ICB in OSCC treatment [27]. In addition, pyroptosis could stimulate innate immunity and induce the release of cytokines (such as IL-1β) (Supplementary Fig. 5) and other molecules (such as HMGB1 (high mobility group box 1) and calreticulin (CRT)) (Supplementary Fig. 6) after cell membrane rupture, form cell immunogenic death, thereby trigger immune effects.
In this work, an orthotopic OSCC tumor model was built by oral administration of 4-NQO to C57BL/6 mice and utilized to evaluate the role of pyroptosis in OSCC. All mice were sacrificed after treatments (Fig. 3a). Figure 3b showed the tongue tissue images of each group after treatment; Fig. 3c, d summarized the average OSCC numbers and areas in each group. TUNEL staining of tissues indicated the death cells (Fig. 3e). In DAC + DDP group, more dead cells appeared, indicated improved antitumor efficacy.

Pretreatment with DAC combined with DDP and anti-PD-L1 demonstrated improved antitumor efficacy. A Timeline of 4-NQO induction the orthotopic OSCC mice model and treatment schedules; B Images of different treatments for tongue lesions, red areas are lesions (scale bar = 3 mm); C, D Mean number and areas of lesions per OSCC mice after different treatment; E Dead tumor cell detected by TUNEL staining (scale bar = 200 μm)
Next, peripheral bloods, tumor-infiltrating lymphocytes (TIL) and cervical lymph nodes in each group were collected for flow cytometry analysis to test the proportion of different lymphocytes in peripheral blood, including CD4+ T, CD8+ T, B, NK, MDSC, macrophage and Treg cells (Fig. 4a, c). Among TILs, the maturation of DCs (CD80+, CD86+) and killer markers of CD8+ T (IFN-γ, TNF-α, IL-2, GzmB) were measured (Fig. 4b, d). Cervical lymph nodes were collected to detect the differentiation of T cells (Fig. 4e, f; CD44+CD62L+ (Tcm); CD44+CD62L− (Teff); CD44−CD62L+ (Tn); CD44−CD62L− (Tem)). These results showed that proportion of CD8+ T cells greatly increased in the mice treated by the combination of DAC and chemodrugs. In cervical lymph nodes, increasing in the proportion of primary T cells in combined with ICB treatments group indicated an enhanced capacity of T cells to activate, proliferate and differention into effector and memory CD8+ T cells. It was evident in the results of mIHC that as the drug combination increased, the proportions of CD8+ T cells, CD11c and cleaved-caspase3 also gradually increased (Fig. 4g). In summary, these results demonstrated that DAC pretreatment combined with chemotherapy and immunotherapy could achieve the best therapeutic effect, mainly due to the change of the suppressive TME of OSCC after triggering pyroptosis (Fig. 4h).

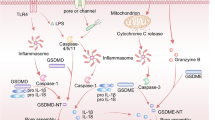
Pyroptosis of tumor cells enhanced the immune response to chemotherapy and cytotoxicity function of CD8+ T cells. A, C tSNE showed the proportion of lymphocytes in different treatment groups and statistical results; B, D the mean proportion of IFN-γ, TNF-α, IL-2 and GzmB in CD8+ TIL in different treatment groups and statistical results displayed by tSNE; E, F flow cytometry presentment the differentiation and statistics of CD8+ T cells in cervical lymph nodes; G mIHC displayed the functionality marker of CD8 (green), CD11c (red) and cleaved-caspase3 (yellow)(scale bar = 200 μm), white arrows showed the positive cells in different group. H Schematic of GSDME regulating OSCC "cold tumor" to "hot tumor" after reversed expression by methyltransferase inhibitor (Color figure online)
GSDME enhances the functional properties of immune cells
Overexpression of GSDME could enhance the sensitivity of OSCC cells to chemotherapy, trigger pyroptosis of tumor cells (Fig. 2). Pretreatment of DAC could induce GSDME higher expression than untreated cells, improving the therapeutic efficiency and anti-tumor immune response in vitro and in vivo (Fig. 3, 4). However, the mechanism of GSDME inducing pyroptosis in OSCC is still unclear. Next, we studied the mechanism of GSDME in OSCC.
Figure 5a is the scheme of induce an orthotopic OSCC mouse model. 14/20 tumors were formed OSCC in WT mice, while 18/20 tumors were formed OSCC in KO mice (Fig. 5b). HE staining showed that WT mice had only moderate or severe epithelial cell proliferation, but tumor cell infiltration and appearance of cancer nests occurred in KO mice (Fig. 5c), indicating that the loss of GSDME would aggravate the pathological condition of 4-NQO induced OSCC model.

GSDME enhances the functional properties of immune cells. A 4-NQO induction scheme of OSCC mice model in WT/KO mice; B number of WT/KO OSCC mice induced successfully; C HE staining of lesions in WT/KO OSCC mice (scale bar = 200 μm); D, E Proportion of lymphocytes in WT/KO OSCC mice displayed by tSNE; F–G Proportion of IFN-γ, TNF-α, IL-2 and GzmB in CD8+ T cells in WT/KO OSCC mice showed by tSNE; H BMDCs mature ability in WT/KO mice; I, BMDCs from WT/KO mice were co-cultured with CD8+ T cells for 24 h to examine their maturation ability (CD80+CD86.+)
GSDME inhibits tumor growth by increasing the antitumor function of tumor-infiltrating NK cells and CD8+ T killer cells in melanoma [15]. For untreated WT and KO mice, the immune system was normal and there was no difference in the proportion of lymphocytes [13]. However, after OSCC induction, the proportion and function of lymphocytes in peripheral blood of mice were detected and significant differences were found in CD4+ T, CD8+ T, NK, DC, MDSC, macrophage and Treg cells (Fig. 5d, e). Tumor expressing GSDME enhanced immune function suggests that tumor suppression may be immune-mediated. This has been shown in our previous studies (Fig. 3, 4). We further investigated the effect of GSDME on peripheral blood CD8+ T cells to evaluate whether T lymphocytes were involved in the improvement process. The level of GzmB, IFN-γ and IL-2 production by CD8+ T cells in the WT group were higher compared with KO group, while no significant difference of TNF-α production was found between the two groups (Fig. 5f, g). These results suggested that GSDME enhanced the effector function of CD8+ T cells in vivo.
Next, we explored how GSDME expression affected the function of CD8+ T lymphocytes. DC-T cell functional interactions are the key to the generation of antitumor immunity. DC has long been recognized as a professional antigen presenting cell that initiates CD4+ or CD8+ T cells [5]. We obtained the Bone Marrow-Derived Dendritic Cells (BMDCs) from WT and KO mice, respectively and induced their maturation in vitro, examined their mature ability and found that the BMDCs mature ability of KO mice were lower than WT mice (Fig. 5h). In addition, WT CD8+ T cells by flow sorting and co-cultured with WT or KO BMDCs for 24 h respectively and test the maturation of BMDCs (Fig. 5i), proliferation of CD8+ T cells (Supplementary Fig. 7) and CD8+ T cell immune function (Supplementary Fig. 8), results showed that KO mice had weaker proliferative ability and anti-tumor immune function compared with WT mice.
Discussion
In the present work, we first demonstrated that DDP, a clinical first-line therapy for OSCC, could induce moderate pyroptosis in HN6 and cal27. This process dependent on GSDME/caspase3 pathway. Pretreatment of methyltransferase inhibitor DAC could induce the expression of GSDME and effective trigger pyrotposis when combined with DDP. In addition, combination with immunotherapy showed promising antitumor results in vivo. This new combination therapy strategy, which can overcome the problem of low response rate of OSCC to anti-PD-L1. Pro-inflammatory pyroptosis recruited more immune cells to tumor sites, reverse OSCC from “cold” tumor to “hot” tumor and improve response of ICB (Fig. 4H). Next, we explored the functional mechanism of GSDME as a tumor suppressor in OSCC. Inducing the orthotopic OSCC model in WT/KO mice, we found that CD8+ T cells in KO mice could not effectively activated as well as WT mice. Subsequently, we explored that it was due to the immaturity of BMDCs in KO mice that could not effectively present antigens to activate CD8+ T cells (Fig. 5). This study provided preclinical treatment references for OSCC and proposed a new functional strategy for pyroptosis in OSCC therapy.
As the executor of pyroptosis, the expression of GSDME in many tumor cells was lower than that in normal cells, due to abnormal methylation of GSDME in tumor cells [28]. Methylation imbalance is one of the characteristics of most malignancies. Studies had shown that methylation modifications of genetic molecules were involved in the regulation of a variety of tumor-related pathways [29]. In addition, in order to prove that amplified pyroptosis after DAC was responsible for reversing GSDME silencing, we constructed a sh-GSDME plasmid and found that the proportion of pyroptosis decreased after using the same treatment, suggesting that amplified pyroptosis response by pretreatment of DAC combined with chemotherapeutic was mainly due to reversal of GSDME expression.
Apoptosis and necrosis are the first found two death types of cell death, which have different effects on inflammation and immunity [30]. In the process of apoptosis, cell membrane shrinkage, apoptotic bodies form and phagocytosed by the surrounding macrophages, it is a non-inflammatory death [31]. In necrosis, membrane bursts and the contents exude, causing the surrounding immune response, it is a pro-inflammatory death [32]. Unlike apoptosis, pyroptosis is a kind of programmed cell death with high pro-inflammatory activity. The gasdermin family is the key protein in pyrotosis execution. Cutting gasdermin by different caspases could trigger pyroptosis in cancer cells [33]. Caspase3 is a co-executor of GSDME associated pyroptosis. Transition from apoptosis to pyroptosis seems to be a promising cancer treatment strategy. Among the tumor cells, GSDME high expression cells were pyroptosis when treated with chemotherapeutic drugs, while GSDME low expression cells underwent apoptosis [34]. We demonstrated that DDP treatment could enhance the pyroptosis ratio when GSDME overexpression (Fig. 2). Inhibition of caspase3 by specific inhibitor Z-DEVD-FMK, could prevent the activation of GSDME and pyroptosis of OSCC cells, implying that caspase3 is involved in the pyroptosis of OSCC cells through the cleavage of GSDME (Fig. 1).
Cancer arises from long-term inflammation, and inflammatory cytokines induce tumor cell necrosis and pyroptosis, which may promote the occurrence of tumors [35]. Tumor could recruit cytokines and chemokines released by leukocytes, which accelerate tumor growth [36]. Literatures have shown that TAMs and MDSCs are often associated with inhibition of anti-tumor immunity and tumor progression, predict poor prognosis of cancer patients [37]. Previous reports have shown that GSDME mediates tumor pyroptosis in vivo by direct lysis of GzmB released from killer lymphocytes, which increases the infiltration of effector lymphocytes (including macrophages, NK cells, and CD8+ T cells). This positive feedback loop is dependent on pyroptosis induced recruitment of killer lymphocytes [15]. In our study, we found that GSDME−/− mice reduced infiltration of immune cells and decreased function of CD8+ T cells in the presence of OSCC mice model. In addition, BMDCs from GSDME−/− mice had decreased antigen presentation ability and could not activate CD8+ T cells to exert cytotoxic functions. This should be a new finding between GSDME and immune function, how GSDME affects immune system is also our next step works.
In summary, our study here demonstrated that stimulation of GSDME expression could improve the sensitivity of chemotherapeutics, activate inflammatory tumor cell pyroptosis and alter the tumor immune-suppressive microenvironment. Our investigation opens new thoughts for future cancer treatment to improve anti-tumor immunity by promoting inflammatory pyroptosis. Activation of pyroptosis of tumor cells and reverse of tumor immunosuppressive microenvironment promising therapeutic outcomes value for OSCC patients.
Data Availability
The data sets used and analyzed in the current study are available from the corresponding author on reasonable request. All data generated or analyzed during this study are included in this published article and its supplementary files.
Abbreviations
- OSCC:
-
Oral squamous cell carcinoma
- TME:
-
Tumor microenvironment
- ICD:
-
Immunogenic cell death
- DAC:
-
Decitabine
- DDP:
-
Cisplatin
- HNSCC:
-
Head and neck squamous cell carcinoma
- GSDME:
-
Gasdermin E
References
Chow LQM (2020) Head and neck cancer. N Engl J Med 382(1):60–72
Johnson D, Burtness B, Leemans C, Lui V, Bauman J, Grandis J (2020) Head and neck squamous cell carcinoma. Nat Rev 6(1):92
Vermorken J, Mesia R, Rivera F et al (2008) Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Eng J Med 359(11):1116–1127
Binnewies M, Roberts E, Kersten K et al (2018) Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 24(5):541–550
Duan Q, Zhang H, Zheng J, Zhang L (2020) Turning cold into hot: firing up the tumor microenvironment. Trends Cancer 6(7):605–618
Peltanova B, Raudenska M, Masarik M (2019) Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma: a systematic review. Mole Cancer 18(1):63
Saddawi-Konefka R, Simon A, Sumner W, Sharabi A, Mell L, Cohen E (2021) Defining the role of immunotherapy in the curative treatment of locoregionally advanced head and neck cancer: promises, challenges, and opportunities. Frontiers Oncol 11:738626
Mei Z, Huang J, Qiao B, Lam AK (2020) Immune checkpoint pathways in immunotherapy for head and neck squamous cell carcinoma. Int J Oral Sci 12(1):16
Dai X, Wang D, Zhang J (2021) Programmed cell death, redox imbalance, and cancer therapeutics. Apoptosis 26:385–414
Zhang Y, Chen X, Gueydan C, Han J (2018) Plasma membrane changes during programmed cell deaths. Cell Res 28(1):9–21
Van Laer L, Huizing E, Verstreken M et al (1998) Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Gene 20(2):194–197
Erkes DA, Cai W, Sanchez IM et al (2020) Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Discov 10(2):254–269
Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F (2017) Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547(7661):99–103
Wang S, Zhang MJ, Wu ZZ et al (2022) GSDME is related to prognosis and response to chemotherapy in oral cancer. J Dental Res 101(7):848–858
Zhang Z, Zhang Y, Xia S et al (2020) Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579(7799):415–420
Saeki N, Kuwahara Y, Sasaki H, Satoh H, Shiroishi T (2000) Gasdermin (Gsdm) localizing to mouse Chromosome 11 is predominantly expressed in upper gastrointestinal tract but significantly suppressed in human gastric cancer cells. Mamma Geno 11(9):718–724
Yu J, Li S, Qi J, Chen Z, Wu Y, Guo J, Wang K, Sun X, Zheng J (2019) Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Dea Dise 10(3):193
Adelstein D, Gillison M, Pfister D et al (2017) NCCN guidelines insights: head and neck cancers, version 2.2017. J Nat Compre Cancer Net 15(6):761–70
Gal-Yam E, Saito Y, Egger G, Jones P (2008) Cancer epigenetics: modifications, screening, and therapy. Annu Rev Med 59:267–80
Akino K, Toyota M, Suzuki H et al (2007) Identification of DFNA5 as a target of epigenetic inactivation in gastric cancer. Cancer Sci 98(1):88–95
Kim M, Chang X, Yamashita K et al (2008) Aberrant promoter methylation and tumor suppressive activity of the DFNA5 gene in colorectal carcinoma. Oncogene 27(25):3624–3634
Wijermans P, Lübbert M, Verhoef G, Bosly A, Ravoet C, Andre M, Ferrant A (2000) Low-dose 5-aza-2’-deoxycytidine, a DNA hypomethylating agent, for the treatment of high-risk myelodysplastic syndrome: a multicenter phase II study in elderly patients. J Clin Oncol 18(5):956–962
Silverman L, Demakos E, Peterson B et al (2002) Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol 20(10):2429–2440
Pardoll D (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev 12(4):252–264
Wise-Draper T, Gulati S, Palackdharry S et al (2022) Phase II clinical trial of neoadjuvant and adjuvant pembrolizumab in resectable local-regionally advanced head and neck squamous cell carcinoma. Clin Cancer Res 28(7):1345–1352
Saada-Bouzid E, Peyrade F, Guigay J (2019) Immunotherapy in recurrent and or metastatic squamous cell carcinoma of the head and neck. Curr Opin Oncol 31(3):146–151
Ferris R, Blumenschein G, Fayette J et al (2016) Nivolumab for recurrent squamous-cell carcinoma of the head and neck. New Engl J Med 375(19):1856–1867
Ibrahim J, Op de Beeck K, Fransen E, Croes L, Beyens M, Suls A, Vanden Berghe W, Peeters M, Van Camp G (2019) Methylation analysis of Gasdermin E shows great promise as a biomarker for colorectal cancer. Cancer Med 8(5):2133–2145
Souza CF, Sabedot TS, Malta TM, Stetson L, Morozova O, Sokolov A et al (2018) A distinct DNA methylation shift in a subset of glioma CpG island methylator phenotypes during tumor recurrence. Cell Rep 23(2):637–651
Wallach D, Kang T, Kovalenko A (2014) Concepts of tissue injury and cell death in inflammation: a historical perspective. Nature Rev Immunol 14(1):51–59
Nowak-Sliwinska P, Griffioen AW (2018) Apoptosis on the move. Apoptosis 23(5–6):251–254
Wallach D, Kang TB, Dillon CP, Green DR (2016) Programmed necrosis in inflammation: toward identification of the effector molecules. Science 352(6281):aaf2154
Kesavardhana S, Malireddi RKS, Kanneganti TD (2020) Caspases in cell death, inflammation, and pyroptosis. Annu Rev Immunol 38:567–95
Lu H, Zhang S, Wu J, Chen M, Cai MC (2018) Molecular targeted therapies elicit concurrent apoptotic and GSDME-dependent pyroptotic tumor cell death. Clin Cancer Res 24(23):6066–6077
Philip M, Rowley DA, Schreiber H (2004) Inflammation as a tumor promoter in cancer induction. Semin Cancer Biol 14(6):433–439
Vakkila J, Lotze MT (2004) Inflammation and necrosis promote tumour growth. Nature Rev Immunol 4(8):641–648
Ma S, Song W, Xu Y, Si X, Zhang Y, Tang Z, Chen X (2020) A ROS-responsive aspirin polymeric prodrug for modulation of tumor microenvironment and cancer immunotherapy. CCS Chem 2(6):390–400
Acknowledgements
The authors thank the National Center for Protein Science at Peking University in Beijing, China, for assistance with Nikon A1R confocal microscopy photography and instruction of using flow cytometer. We thank Su Lab’s help in the cell experiments at Biomedical Pioneering Innovation Center, State Key Laboratory of Protein and Plant Gene Research, School of Life Science, Peking University. We thank for Figdraw website for the picture drawing.
Funding
This work was supported by Discipline Development Fund of School of Stomatology, Peking University.
Author information
Authors and Affiliations
Contributions
ZM: contributed to conceptualization, methodology, data analysis, writing original draft, writing, review and editing; XYC: contributed to conceptualization, methodology, data analysis; YC: contributed to data analysis; XDS: contributed to original draft review; SXL: contributed to conceptualization, funding acquisition, supervision, writing-review and editing; SCW: contributed to supervision, project administration, writing-review and editing. All authors gave final approval and agree to be accountable for all aspects of the work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical Approval
This study was performed in line with the principles of the Medical Ethics Committee and Biosafety Management Committee of Peking University (Approval Number LA2019197).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zi, M., Xingyu, C., Yang, C. et al. Improved antitumor immunity of chemotherapy in OSCC treatment by Gasdermin-E mediated pyroptosis. Apoptosis 28, 348–361 (2023). https://doi.org/10.1007/s10495-022-01792-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-022-01792-3