
Abstract
Similar to apoptosis of nucleated cells, erythrocytes may undergo eryptosis, a suicidal death characterized by cell shrinkage and phospholipid scrambling of the cell membrane leading to phosphatidylserine exposure at the cell surface. As eryptotic erythrocytes are rapidly cleared from circulating blood, excessive eryptosis may lead to anemia. Moreover, eryptotic erythrocytes may adhere to the vascular wall and thus impede microcirculation. Stimulators of eryptosis include osmotic shock, oxidative stress and energy depletion. Mechanisms involved in the stimulation eryptosis include ceramide formation which may result from phospholipase A2 dependent formation of platelet activating factor (PAF) with PAF dependent stimulation of sphingomyelinases. Enhanced erythrocytic ceramide formation is observed in fever, sepsis, HUS, uremia, hepatic failure, and Wilson’s disease. Enhanced eryptosis is further observed in iron deficiency, phosphate depletion, dehydration, malignancy, malaria, sickle-cell anemia, beta-thalassemia and glucose-6-phosphate dehydrogenase-deficiency. Moreover, eryptosis is triggered by osmotic shock and a wide variety of xenobiotics, which are again partially effective by enhancing ceramide abundance. Ceramide formation is inhibited by high concentrations of urea. As shown in Wilson’s disease, pharmacological interference with ceramide formation may be a therapeutic option in the treatment of eryptosis inducing clinical disorders.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Mature erythrocytes eventually undergo senescence leading to their removal from circulation within 100–120 days [1–3]. Erythrocyte senescence involves binding of hemichromes to band 3 with subsequent band 3 clustering, deposition of complement C3 fragments and binding of anti-band 3 immunoglobulins [4].
Erythrocytes may face injury prior to senescence. Defective erythrocytes may eventually undergo hemolysis and release hemoglobin, which may be filtered in renal glomeruli and subsequently precipitate in the acidic lumen of renal tubules [5]. To avoid hemolysis, erythrocytes may enter suicidal death or eryptosis, which is characterized by cell shrinkage and cell membrane scrambling [6–9]. The cell shrinkage serves to counteract cell swelling, the phosphatidylserine exposure fosters phagocytosis [10, 11] and thus clearance of affected erythrocytes from circulating blood [12].
Excessive stimulation of eryptosis, however, causes anemia as soon as the clearance of eryptotic erythrocytes from circulating blood surpasses the formation of new erythrocytes [6–9]. Phosphatidylserine exposing erythrocytes further adhere to the vascular wall [9] and blood platelets [13], trigger blood clotting and thrombosis and interfere with microcirculation [9]. Thus, enhanced eryptosis may become pathophysiologically relevant. Eryptosis is distinct from programmed erythrocyte necrosis [14].
Mechanisms involved in the triggering of eryptosis include ceramide formation. The present review briefly discusses the cellular mechanisms involved in the triggering of eryptosis with particular emphasis on ceramide. It further discusses the contribution of ceramide to the triggering of eryptosis in disease and by xenobiotics. The reader is encouraged to consult earlier, more extensive reviews providing the description of further aspects of eryptosis [6–8, 15–18].
Triggers and clinical conditions associated with enhanced eryptosis
A wide variety of chemicals triggers eryptosis [18–31]. Moreover, enhanced eryptosis is observed in several disorders including iron deficiency [12], phosphate depletion [32], dehydration [33], Parkinson’s disease [34], fever [35], sepsis [36], hemolytic anemia [37], hemolytic uremic syndrome (HUS) [38], end stage renal disease [39–41], metabolic syndrome [42], diabetes [43–45], hepatic failure [46], malignancy [47], malaria [7, 48–52], sickle-cell disease [15, 50, 51, 53–56], thalassemia [51, 53, 55, 57–59], glucose-6-phosphate dehydrogenase (G6PD)-deficiency [55, 60], Wilson’s disease [17], mutation or lack of the anion exchanger [61] and an extremely rare mutation of GLUT1 turning the carrier into a Ca2+ permeable cation channel [62]. Enhanced suicidal erythrocyte death is further observed following return from high altitude, a phenomenon affecting mainly newly formed erythrocytes (neocytolysis) [63].
Role of calcium in eryptosis
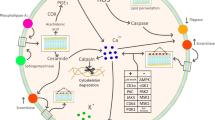
Eryptosis is stimulated by increase in cytosolic Ca2+ activity [64–66], which is known to trigger vesiculation of the cell membrane [67] and cell membrane scrambling [45, 68, 69]. Increased cytosolic Ca2+ activity further activates the cysteine endopeptidase calpain, an enzyme degrading the cytoskeleton and thus leading to cell membrane blebbing [70]. Beyond that, an increase of cytosolic Ca2+ activity is followed by stimulation of Ca2+-sensitive K+ channels [71–73] with subsequent K+ efflux, hyperpolarization of the cell membrane, Cl− exit [74] and thus cell shrinkage due to loss of cellular KCl with osmotically obliged water [74]. Mechanisms increasing cytosolic Ca2+ activity include Ca2+ entry through non-selective cation channels [75–79]. The molecular identity of the cation channels is still ill defined but apparently involves TRPC6 [80]. The Ca2+ permeable cation channels are stimulated by osmotic shock [81, 82], oxidative stress [82, 83] and Cl− removal [79, 81, 83].
Oxidative stress and further signaling triggering eryptosis
Oxidative stress [55, 84, 85] or impaired antioxidative defence [86–88] triggers eryptosis. Oxidative stress is not only effective by activating the Ca2+-permeable cation channels [83] but, in addition activates erythrocyte Cl− channels [89, 90], which contribute to eryptotic cell shrinkage [91]. Oxidative stress further stimulates eryptosis by activation of caspases [65, 92, 93].
The signalling governing eryptosis involves several kinases [18]. Eryptosis is fostered by activation of casein kinase 1α, Janus-activated kinase JAK3, protein kinase C, and p38 kinase [18]. Eryptosis is inhibited by AMP activated kinase AMPK, cGMP-dependent protein kinase, PAK2 kinase, sorafenib sensitive kinases and sunifinib sensitive kinases [18].
Gene targeted mice with enhanced eryptosis include mice lacking AMP-activated protein kinase [94], cGMP-dependent protein kinase [95], Klotho [96], or AE1 [97]. Moreover, enhanced eryptosis is observed in mice expressing excessive erythropoietin levels [98]. Decreased eryptosis is observed in mice deficient in PAF receptor [99], PDK1 [100] or TRPC6 [80]. The susceptibility to eryptosis increases with erythrocyte age [101, 102].
Sphingomyelin breakdown and ceramide formation
A major stimulator of eryptosis is ceramide [103, 104], which has previously been shown to participate in the triggering of apoptosis of a wide variety of cell types [105–111]. Sphingolipids consist of an sphingoid base, i.e. a 1,3-dihydroxy-2-aminoalken backbone with Sphingosine, i.e. (2S, 3R, 4E)-2-amino-4-octadecene-1,3-diol, being the most prevalent backbone of mammalian sphingolipids. The attachment of sphingosine to a fatty acid via an amide ester bond results in the formation of ceramide [112, 113]. Ceramides contain fatty acids with very different chain lengths from 2 to 36 carbon atoms in the acyl chain and also differ in their saturation. Modification of ceramide by attachments of various headgroups results—for instance—in sphingomyelin, gangliosides, sulfatides, globosides or cerebrosides [112, 113]. Sphingomyelin, an ester of a ceramide moiety and a hydrophilic phosphorylcholine headgroup, is the most prevalent sphingolipid in the cell membrane [112, 113]. Ceramide is mainly generated via a de novo synthesis pathway or by hydrolysis of sphingomyelin, a step catalyzed by sphingomyelinases [for review see [114]]. Under certain circumstances ceramide is also generated by a retrograde activity of ceramidases converting sphingosine into ceramide or by hydrolysis of complex glycosylated lipids or ceramide 1-phosphate [for review see [114]]. Sphingomyelinases cleave phospho-diester bonds and belong to the family of hydrolases. The pH value, but also the composition of the membrane, determines the activity of sphingomyelinases [115, 116] and, therefore, sphingomyelinases are classified into an acid and several neutral and alkaline sphingomyelinases. The acid sphingomyelinase functions best at a pH of 4.5–5.0, but the lipid composition of the membrane alters the Km of the enzyme and thereby acid sphingomyelinase also functions at higher pH for instance at plasma membranes [116]. Acid and neutral sphingomyelinases are the enzymes, which are mainly involved in eryptosis as evidenced by genetic and pharmacological studies using functional inhibitors of the acid sphingomyelinase such as amitriptyline or imipramine [104, 117–119].
Besides the hydrolysis of sphingomyelin, ceramide is generated de novo by the activity of several enzymes including serine palmitoyl-transferase and ceramide synthases. The serine palmitoyl-transferase catalyzes the condensation of l-serine and palmitoyl CoA to 3-ketosphinganine, which is the rate-limiting enzyme in sphingolipid biosynthesis. 3-ketosphinganine is reduced to sphinganine by a reductase. The N-acylation of sphinganine to dihydro-ceramide is catalyzed by ceramide synthases. Finally, dihydro-ceramide is desaturated by desaturase to ceramide [120]. At present six ceramide synthases, i.e. ceramide synthase 1–6, are cloned. The ceramide synthases employ acyl-CoA of distinct length to generate (dihydro) ceramide in the de novo biosynthesis pathway and the specificity of each ceramide synthase is limited to a certain chain length, for instance CerS1 uses mostly C18-fatty acyl CoA [121], CerS2 can utilize a wider range of very long chain (VLC) fatty acyl CoAs (C20 to C26) [122]. CerS3 incorporates ultra-long chain fatty acyl CoAs (C26 to C32) [123, 124], CerS4 uses C18- and C20-fatty acyl CoAs [125]. CerS5 has specificity only for C16-fatty acyl CoA [123], and CerS6 can use both C14- and C16-fatty acyl CoAs [126].
Role of ceramide in eryptosis and adhesion to the vascular wall
In erythrocytes ceramide is generated following osmotic shock by sphingomyelin breakdown [103]. Ceramide sensitizes erythrocytes to the eryptotic effect of enhanced Ca2+ concentration [103]. Along those lines erythrocyte cell membrane scrambling following osmotic shock is mimicked by addition of C6-ceramide, C16-ceramide or addition of bacterial sphingomyelinase [103].
The mechanisms involved in ceramide induced eryptosis remained incompletely understood. In nucleated cells ceramide fosters receptor clustering in lipid rafts and formation of a death-inducing signalling complex (DISC) [127–130], modifies the membrane curvature and thus compromises cell membrane integrity [131, 132]. In erythrocytes ceramide is similarly localized in clusters [133]. Ceramide modifies the interaction of the membrane with the cytoskeleton and increases membrane fragility [133]. Eventually, ceramide-induced changes in the membrane lead to vesiculation, rigidity and enhanced membrane permeability [133].
Enhanced ceramide abundance is involved in the triggering of eryptosis by fever [35], sepsis [36], hemolytic anemia [37], HUS [38], end stage renal disease [40], hepatic failure [46], and Wilson’s disease [17]. Table 1 lists various xenobiotics triggering eryptosis at least in part by increasing the ceramide abundance. Ceramide formation is inhibited by amitriptyline [117, 134] and urea [135]. The enzyme accomplishing the formation of ceramide has, however, remained ill defined. In sepsis [36], HUS [38] and end stage renal disease [40], the eryptosis could be triggered by exposure of erythrocytes from healthy individuals to patient plasma. In theory, the plasma could harbour a ceramide-producing enzyme such as sphingomyelinase in those diseases. Sphingomyelinase activity has indeed been detected in the serum of patients suffering from Wilson’s disease [17]. In erythrocytes, ceramide formation could be triggered by platelet-activating factor PAF [99]. Osmotic erythrocyte shrinkage triggers PAF formation by a phospholipase A2 [99]. Erythrocytes express PAF receptors at the erythrocyte surface and exposure of erythrocytes to PAF stimulates sphingomyelin breakdown and ceramide formation, effects disrupted by genetic knockout of the PAF receptor [99]. Exposure of erythrocytes to bacterial sphingomyelinase triggers eryptosis and subsequent adhesion of the erythrocytes to endothelial cells [136]. Adhesion is blunted by phosphatidylserine-coating annexin-V, by addition of neutralizing antibodies against endothelial CXCL16 and by silencing of the CXCL16 gene with small interfering RNA. Pretreatment of the endothelial cells with bacterial sphingomyelinase upregulates CXCL16 protein abundance thus fostering adhesion not only by triggering of eryptosis but as well by enhancing docking molecules at the endothelial surface [136]. Transmembrane CXCL16 serves as an endothelial adhesion molecule not only for eryptotic cells but as well for lymphocytes [137]. CXCL16 expression is enhanced in atherosclerotic lesions [137, 138]. It is tempting to speculate that the preferred CXCL16 expression in atherosclerotic plaques could foster recruitment of eryptotic erythrocytes to those sites, which could contribute to the development of thrombosis.
Conclusions
Ceramide formation participates in the stimulation of eryptosis, the suicidal erythrocyte death. Several diseases and a wide variety of xenobiotics stimulate eryptosis at least in part by increasing ceramide abundance. Ceramide formation is stimulated by PAF, which is generated by a phospholipase A2. Additional experiments are required to define the ceramide-generating enzyme(s) and the molecular mechanisms involved in ceramide-dependent cell membrane scrambling.
Abbreviations
- AE1:
-
Anion exchanger 1
- AMP:
-
Adenosinmonophosphate
- AMPK:
-
AMP activated kinase
- GMP:
-
Cyclic guanosinmonophosphate
- CXCL16:
-
CXC-Motiv-Chemokin 16
- HUS:
-
Hemolytic uremic syndrome
- G6PD:
-
Glucose-6-phosphate dehydrogenase
- GLUT1:
-
Glucose transporter 1
- PAF:
-
Platelet activating factor
- PAK2:
-
p21-activated kinase 2
- PDK1:
-
Phosphoinositide dependent kinase 1
- TRPC6:
-
Transient receptor potential channel C6
References
Arese P, Turrini F, Schwarzer E (2005) Band 3/complement-mediated recognition and removal of normally senescent and pathological human erythrocytes. Cell Physiol Biochem 16:133–146
Bosman GJ, Willekens FL, Werre JM (2005) Erythrocyte aging: a more than superficial resemblance to apoptosis? Cell Physiol Biochem 16:1–8
Kiefer CR, Snyder LM (2000) Oxidation and erythrocyte senescence. Curr Opin Hematol 7:113–116
Lutz HU (2004) Innate immune and non-immune mediators of erythrocyte clearance. Cell Mol Biol (Noisy-le-grand) 50:107–116
Harrison HE, Bunting H, Ordway NK, Albrink WS (1947) The pathogenesis of the renal injury produced in the dog by hemoglobin or methemoglobin. J Exp Med 86:339–356
Cimen MY (2008) Free radical metabolism in human erythrocytes. Clin Chim Acta 390:1–11
Foller M, Bobbala D, Koka S, Huber SM, Gulbins E, Lang F (2009) Suicide for survival–death of infected erythrocytes as a host mechanism to survive malaria. Cell Physiol Biochem 24:133–140
Lang F, Gulbins E, Lerche H, Huber SM, Kempe DS, Foller M (2008) Eryptosis, a window to systemic disease. Cell Physiol Biochem 22:373–380
Lang E, Qadri SM, Lang F (2012) Killing me softly—suicidal erythrocyte death. Int J Biochem Cell Biol 44:1236–1243
Boas FE, Forman L, Beutler E (1998) Phosphatidylserine exposure and red cell viability in red cell aging and in hemolytic anemia. Proc Natl Acad Sci USA 95:3077–3081
Fadok VA, Bratton DL, Rose DM, Pearson A, Ezekewitz RA, Henson PM (2000) A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 405:85–90
Kempe DS, Lang PA, Duranton C, Akel A, Lang KS, Huber SM, Wieder T, Lang F (2006) Enhanced programmed cell death of iron-deficient erythrocytes. FASEB J 20:368–370
Walker B, Towhid ST, Schmid E, Hoffmann SM, Abed M, Munzer P, Vogel S, Neis F, Brucker S, Gawaz M, Borst O, Lang F (2014) Dynamic adhesion of eryptotic erythrocytes to immobilized platelets via platelet phosphatidylserine receptors. Am J Physiol Cell Physiol 306:C291–C297
LaRocca TJ, Stivison EA, Hod EA, Spitalnik SL, Cowan PJ, Randis TM, Ratner AJ (2014) Human-specific bacterial pore-forming toxins induce programmed necrosis in erythrocytes. MBio 5:e01251–14
Browning JA, Robinson HC, Ellory JC, Gibson JS (2007) Deoxygenation-induced non-electrolyte pathway in red cells from sickle cell patients. Cell Physiol Biochem 19:165–174
Foller M, Sopjani M, Mahmud H, Lang F (2008) Vanadate induced suicidal erythrocyte death. Kidney Blood Pres Res 21:87–93
Lang PA, Schenck M, Nicolay JP, Becker JU, Kempe DS, Lupescu A, Koka S, Eisele K, Klarl BA, Rubben H, Schmid KW, Mann K, Hildenbrand S, Hefter H, Huber SM, Wieder T, Erhardt A, Haussinger D, Gulbins E, Lang F (2007) Liver cell death and anemia in Wilson disease involve acid sphingomyelinase and ceramide. Nat Med 13:164–170
Lang F, Abed M, Lang E, Foller M (2014) Oxidative stress and suicidal erythrocyte death. Antioxid Redox Signal 21:138–153
Bissinger R, Lupescu A, Zelenak C, Jilani K, Lang F (2014) Stimulation of eryptosis by cryptotanshinone. Cell Physiol Biochem 34:432–442
Jacobi J, Lang E, Bissinger R, Frauenfeld L, Modicano P, Faggio C, Abed M, Lang F (2014) Stimulation of erythrocyte cell membrane scrambling by mitotane. Cell Physiol Biochem 33:1516–1526
Lupescu A, Bissinger R, Herrmann T, Oswald G, Jilani K, Lang F (2014) Induction of suicidal erythrocyte death by novobiocin. Cell Physiol Biochem 33:670–680
Lupescu A, Bissinger R, Warsi J, Jilani K, Lang F (2014) Stimulation of erythrocyte cell membrane scrambling by gedunin. Cell Physiol Biochem 33:1838–1848
Lupescu A, Jilani K, Zbidah M, Lang F (2013) Patulin-induced suicidal erythrocyte death. Cell Physiol Biochem 32:291–299
Tesoriere L, Attanzio A, Allegra M, Cilla A, Gentile C, Livrea MA (2014) Oxysterol mixture in hypercholesterolemia-relevant proportion causes oxidative stress-dependent eryptosis. Cell Physiol Biochem 34:1075–1089
Aguilar-Dorado IC, Hernandez G, Quintanar-Escorza MA, Maldonado-Vega M, Rosas-Flores M, Calderon-Salinas JV (2014) Eryptosis in lead-exposed workers. Toxicol Appl Pharmacol 281:195–202
Gao M, Lau PM, Kong SK (2014) Mitochondrial toxin betulinic acid induces in vitro eryptosis in human red blood cells through membrane permeabilization. Arch Toxicol 88:755–768
Hoelzle LE, Zeder M, Felder KM, Hoelzle K (2014) Pathobiology of mycoplasma suis. Vet J 202:20–25
Oswald G, Alzoubi K, Abed M, Lang F (2014) Stimulation of suicidal erythrocyte death by ribavirin. Basic Clin Pharmacol Toxicol 114:311–317
Zhang R, Xiang Y, Ran Q, Deng X, Xiao Y, Xiang L, Li Z (2014) Involvement of calcium, reactive oxygen species, and atp in hexavalent chromium-induced damage in red blood cells. Cell Physiol Biochem 34:1780–1791
Malik A, Bissinger R, Jilani K, Lang F (2015) Stimulation of erythrocyte cell membrane scrambling by nystatin. Basic Clin Pharmacol Toxicol 116:47–52
Bissinger R, Malik A, Jilani K, Lang F (2014) Triggering of erythrocyte cell membrane scrambling by salinomycin. Basic Clin Pharmacol Toxicol. doi:10.1111/bcpt.12250; [Epub ahead of print]
Birka C, Lang PA, Kempe DS, Hoefling L, Tanneur V, Duranton C, Nammi S, Henke G, Myssina S, Krikov M, Huber SM, Wieder T, Lang F (2004) Enhanced susceptibility to erythrocyte “apoptosis” following phosphate depletion. Pflugers Arch 448:471–477
Abed M, Feger M, Alzoubi K, Pakladok T, Frauenfeld L, Geiger C, Towhid ST, Lang F (2013) Sensitization of erythrocytes to suicidal erythrocyte death following water deprivation. Kidney Blood Press Res 37:567–578
Pretorius E, Swanepoel AC, Buys AV, Vermeulen N, Duim W, Kell DB (2014) Eryptosis as a marker of Parkinson’s disease. Aging 6:788–819
Foller M, Braun M, Qadri SM, Lang E, Mahmud H, Lang F (2010) Temperature sensitivity of suicidal erythrocyte death. Eur J Clin Invest 40:534–540
Kempe DS, Akel A, Lang PA, Hermle T, Biswas R, Muresanu J, Friedrich B, Dreischer P, Wolz C, Schumacher U, Peschel A, Gotz F, Doring G, Wieder T, Gulbins E, Lang F (2007) Suicidal erythrocyte death in sepsis. J Mol Med 85:273–281
Banerjee D, Saha S, Basu S, Chakrabarti A (2008) Porous red cell ultrastructure and loss of membrane asymmetry in a novel case of hemolytic anemia. Eur J Haematol 81:399–402
Lang PA, Beringer O, Nicolay JP, Amon O, Kempe DS, Hermle T, Attanasio P, Akel A, Schafer R, Friedrich B, Risler T, Baur M, Olbricht CJ, Zimmerhackl LB, Zipfel PF, Wieder T, Lang F (2006) Suicidal death of erythrocytes in recurrent hemolytic uremic syndrome. J Mol Med 84:378–388
Myssina S, Huber SM, Birka C, Lang PA, Lang KS, Friedrich B, Risler T, Wieder T, Lang F (2003) Inhibition of erythrocyte cation channels by erythropoietin. J Am Soc Nephrol 14:2750–2757
Abed M, Artunc F, Alzoubi K, Honisch S, Baumann D, Foller M, Lang F (2014) Suicidal erythrocyte death in end-stage renal disease. J Mol Med 92:871–879
Voelkl J, Alzoubi K, Mamar AK, Ahmed MS, Abed M, Lang F (2013) Stimulation of suicidal erythrocyte death by increased extracellular phosphate concentrations. Kidney Blood Press Res 38:42–51
Zappulla D (2008) Environmental stress, erythrocyte dysfunctions, inflammation, and the metabolic syndrome: adaptations to CO2 increases? J Cardiometab Syndr 3:30–34
Calderon-Salinas JV, Munoz-Reyes EG, Guerrero-Romero JF, Rodriguez-Moran M, Bracho-Riquelme RL, Carrera-Gracia MA, Quintanar-Escorza MA (2011) Eryptosis and oxidative damage in type 2 diabetic mellitus patients with chronic kidney disease. Mol Cell Biochem 357:171–179
Maellaro E, Leoncini S, Moretti D, Del Bello B, Tanganelli I, De Felice C, Ciccoli L (2013) Erythrocyte caspase-3 activation and oxidative imbalance in erythrocytes and in plasma of type 2 diabetic patients. Acta Diabetol 50:489–495
Nicolay JP, Schneider J, Niemoeller OM, Artunc F, Portero-Otin M, Haik G Jr, Thornalley PJ, Schleicher E, Wieder T, Lang F (2006) Stimulation of suicidal erythrocyte death by methylglyoxal. Cell Physiol Biochem 18:223–232
Lang E, Gatidis S, Freise NF, Bock H, Kubitz R, Lauermann C, Orth HM, Klindt C, Schuier M, Keitel V, Reich M, Liu G, Schmidt S, Xu HC, Qadri SM, Herebian D, Pandyra AA, Mayatepek E, Gulbins E, Lang F, Haussinger D, Lang KS, Foller M, Lang PA (2015) Conjugated bilirubin triggers anemia by inducing erythrocyte death. Hepatology 61:275–284
Qadri SM, Mahmud H, Lang E, Gu S, Bobbala D, Zelenak C, Jilani K, Siegfried A, Foller M, Lang F (2012) Enhanced suicidal erythrocyte death in mice carrying a loss-of-function mutation of the adenomatous polyposis coli gene. J Cell Mol Med 16:1085–1093
Koka S, Lang C, Boini KM, Bobbala D, Huber S, Lang F (2008) Influence of chlorpromazine on eryptosis, parasitemia and survival of plasmodium berghei infected mice. Cell Physiol Biochem 22:261–268
Koka S, Lang C, Niemoeller OM, Boini KM, Nicolay JP, Huber SM, Lang F (2008) Influence of NO synthase inhibitor L-NAME on parasitemia and survival of Plasmodium berghei infected mice. Cell Physiol Biochem 21:481–488
de Jong K, Emerson RK, Butler J, Bastacky J, Mohandas N, Kuypers FA (2001) Short survival of phosphatidylserine-exposing red blood cells in murine sickle cell anemia. Blood 98:1577–1584
Kean LS, Brown LE, Nichols JW, Mohandas N, Archer DR, Hsu LL (2002) Comparison of mechanisms of anemia in mice with sickle cell disease and beta-thalassemia: peripheral destruction, ineffective erythropoiesis, and phospholipid scramblase-mediated phosphatidylserine exposure. Exp Hematol 30:394–402
Jimenez-Diaz MB, Ebert D, Salinas Y, Pradhan A, Lehane AM, Myrand-Lapierre ME, O’Loughlin KG, Shackleford DM, Justino de Almeida M, Carrillo AK, Clark JA, Dennis AS, Diep J, Deng X, Duffy S, Endsley AN, Fedewa G, Guiguemde WA, Gomez MG, Holbrook G, Horst J, Kim CC, Liu J, Lee MC, Matheny A, Martinez MS, Miller G, Rodriguez-Alejandre A, Sanz L, Sigal M, Spillman NJ, Stein PD, Wang Z, Zhu F, Waterson D, Knapp S, Shelat A, Avery VM, Fidock DA, Gamo FJ, Charman SA, Mirsalis JC, Ma H, Ferrer S, Kirk K, Angulo-Barturen I, Kyle DE, DeRisi JL, Floyd DM, Guy RK (2014) (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. Proc Natl Acad Sci USA 111:E5455–E5462
Ayi K, Turrini F, Piga A, Arese P (2004) Enhanced phagocytosis of ring-parasitized mutant erythrocytes: a common mechanism that may explain protection against falciparum malaria in sickle trait and beta-thalassemia trait. Blood 104:3364–3371
Chadebech P, Habibi A, Nzouakou R, Bachir D, Meunier-Costes N, Bonin P, Rodet M, Chami B, Galacteros F, Bierling P, Noizat-Pirenne F (2009) Delayed hemolytic transfusion reaction in sickle cell disease patients: evidence of an emerging syndrome with suicidal red blood cell death. Transfusion 49:1785–1792
Lang KS, Roll B, Myssina S, Schittenhelm M, Scheel-Walter HG, Kanz L, Fritz J, Lang F, Huber SM, Wieder T (2002) Enhanced erythrocyte apoptosis in sickle cell anemia, thalassemia and glucose-6-phosphate dehydrogenase deficiency. Cell Physiol Biochem 12:365–372
Wood BL, Gibson DF, Tait JF (1996) Increased erythrocyte phosphatidylserine exposure in sickle cell disease: flow-cytometric measurement and clinical associations. Blood 88:1873–1880
Basu S, Banerjee D, Chandra S, Chakrabarti A (2009) Eryptosis in hereditary spherocytosis and thalassemia: role of glycoconjugates. Glycoconj J:in press
Kuypers FA, Yuan J, Lewis RA, Snyder LM, Kiefer CR, Bunyaratvej A, Fucharoen S, Ma L, Styles L, de Jong K, Schrier SL (1998) Membrane phospholipid asymmetry in human thalassemia. Blood 91:3044–3051
Ibrahim HA, Fouda MI, Yahya RS, Abousamra NK, Abd Elazim RA (2014) Erythrocyte phosphatidylserine exposure in beta-thalassemia. Lab Hematol 20:9–14
Cappadoro M, Giribaldi G, O’Brien E, Turrini F, Mannu F, Ulliers D, Simula G, Luzzatto L, Arese P (1998) Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)-deficient erythrocytes parasitized by Plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood 92:2527–2534
Bruce LJ, Robinson HC, Guizouarn H, Borgese F, Harrison P, King MJ, Goede JS, Coles SE, Gore DM, Lutz HU, Ficarella R, Layton DM, Iolascon A, Ellory JC, Stewart GW (2005) Monovalent cation leaks in human red cells caused by single amino-acid substitutions in the transport domain of the band 3 chloride-bicarbonate exchanger, AE1. Nat Genet 37:1258–1263
Weber YG, Storch A, Wuttke TV, Brockmann K, Kempfle J, Maljevic S, Margari L, Kamm C, Schneider SA, Huber SM, Pekrun A, Roebling R, Seebohm G, Koka S, Lang C, Kraft E, Blazevic D, Salvo-Vargas A, Fauler M, Mottaghy FM, Munchau A, Edwards MJ, Presicci A, Margari F, Gasser T, Lang F, Bhatia KP, Lehmann-Horn F, Lerche H (2008) GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak. J Clin Invest 118:2157–2168
Rice L, Alfrey CP (2005) The negative regulation of red cell mass by neocytolysis: physiologic and pathophysiologic manifestations. Cell Physiol Biochem 15:245–250
Berg CP, Engels IH, Rothbart A, Lauber K, Renz A, Schlosser SF, Schulze-Osthoff K, Wesselborg S (2001) Human mature red blood cells express caspase-3 and caspase-8, but are devoid of mitochondrial regulators of apoptosis. Cell Death Differ 8:1197–1206
Bratosin D, Estaquier J, Petit F, Arnoult D, Quatannens B, Tissier JP, Slomianny C, Sartiaux C, Alonso C, Huart JJ, Montreuil J, Ameisen JC (2001) Programmed cell death in mature erythrocytes: a model for investigating death effector pathways operating in the absence of mitochondria. Cell Death Differ 8:1143–1156
Daugas E, Cande C, Kroemer G (2001) Erythrocytes: death of a mummy. Cell Death Differ 8:1131–1133
Allan D, Michell RH (1977) Calcium ion-dependent diacylglycerol accumulation in erythrocytes is associated with microvesiculation but not with efflux of potassium ions. Biochem J 166:495–499
Akel A, Hermle T, Niemoeller OM, Kempe DS, Lang PA, Attanasio P, Podolski M, Wieder T, Lang F (2006) Stimulation of erythrocyte phosphatidylserine exposure by chlorpromazine. Eur J Pharmacol 532:11–17
Niemoeller OM, Akel A, Lang PA, Attanasio P, Kempe DS, Hermle T, Sobiesiak M, Wieder T, Lang F (2006) Induction of eryptosis by cyclosporine. Naunyn Schmiedebergs Arch Pharmacol 374:41–49
Pant HC, Virmani M, Gallant PE (1983) Calcium-induced proteolysis of spectrin and band 3 protein in rat erythrocyte membranes. Biochem Biophys Res Commun 117:372–377
Bookchin RM, Ortiz OE, Lew VL (1987) Activation of calcium-dependent potassium channels in deoxygenated sickled red cells. Prog Clin Biol Res 240:193–200
Brugnara C, de Franceschi L, Alper SL (1993) Inhibition of Ca(2 +)-dependent K + transport and cell dehydration in sickle erythrocytes by clotrimazole and other imidazole derivatives. J Clin Invest 92:520–526
Franco RS, Palascak M, Thompson H, Rucknagel DL, Joiner CH (1996) Dehydration of transferrin receptor-positive sickle reticulocytes during continuous or cyclic deoxygenation: role of KCl cotransport and extracellular calcium. Blood 88:4359–4365
Lang PA, Kaiser S, Myssina S, Wieder T, Lang F, Huber SM (2003) Role of Ca2 + -activated K + channels in human erythrocyte apoptosis. Am J Physiol Cell Physiol 285:C1553–C1560
Bernhardt I, Weiss E, Robinson HC, Wilkins R, Bennekou P (2007) Differential effect of HOE642 on two separate monovalent cation transporters in the human red cell membrane. Cell Physiol Biochem 20:601–606
Ivanova L, Bernhardt R, Bernhardt I (2008) Nongenomic effect of aldosterone on ion transport pathways of red blood cells. Cell Physiol Biochem 22:269–278
Kaestner L, Bernhardt I (2002) Ion channels in the human red blood cell membrane: their further investigation and physiological relevance. Bioelectrochemistry 55:71–74
Kaestner L, Tabellion W, Lipp P, Bernhardt I (2004) Prostaglandin E2 activates channel-mediated calcium entry in human erythrocytes: an indication for a blood clot formation supporting process. Thromb Haemost 92:1269–1272
Lang PA, Kempe DS, Myssina S, Tanneur V, Birka C, Laufer S, Lang F, Wieder T, Huber SM (2005) PGE(2) in the regulation of programmed erythrocyte death. Cell Death Differ 12:415–428
Foller M, Kasinathan RS, Koka S, Lang C, Shumilina E, Birnbaumer L, Lang F, Huber SM (2008) TRPC6 contributes to the Ca(2 +) leak of human erythrocytes. Cell Physiol Biochem 21:183–192
Huber SM, Gamper N, Lang F (2001) Chloride conductance and volume-regulatory nonselective cation conductance in human red blood cell ghosts. Pflugers Arch 441:551–558
Lang KS, Duranton C, Poehlmann H, Myssina S, Bauer C, Lang F, Wieder T, Huber SM (2003) Cation channels trigger apoptotic death of erythrocytes. Cell Death Differ 10:249–256
Duranton C, Huber SM, Lang F (2002) Oxidation induces a Cl(-)-dependent cation conductance in human red blood cells. J Physiol 539:847–855
Barvitenko NN, Adragna NC, Weber RE (2005) Erythrocyte signal transduction pathways, their oxygenation dependence and functional significance. Cell Physiol Biochem 15:1–18
Bracci R, Perrone S, Buonocore G (2002) Oxidant injury in neonatal erythrocytes during the perinatal period. Acta PaediatrSuppl 91:130–134
Bilmen S, Aksu TA, Gumuslu S, Korgun DK, Canatan D (2001) Antioxidant capacity of G-6-PD-deficient erythrocytes. Clin Chim Acta 303:83–86
Damonte G, Guida L, Sdraffa A, Benatti U, Melloni E, Forteleoni G, Meloni T, Carafoli E, De Flora A (1992) Mechanisms of perturbation of erythrocyte calcium homeostasis in favism. Cell Calcium 13:649–658
Mavelli I, Ciriolo M, Rossi L, Meloni T, Forteleoni G, De Flora A, Benatti U, Morelli M, Rotilio G (1984) Favism: a hemolytic disease associated with increased superoxide dismutase and decreased glutathione peroxidase activities in red blood cells. Eur J Biochem 139:8–13
Huber SM, Uhlemann AC, Gamper NL, Duranton C, Kremsner PG, Lang F (2002) Plasmodium falciparum activates endogenous Cl(-) channels of human erythrocytes by membrane oxidation. EMBO J 21:22–30
Tanneur V, Duranton C, Brand VB, Sandu CD, Akkaya C, Kasinathan RS, Gachet C, Sluyter R, Barden JA, Wiley JS, Lang F, Huber SM (2006) Purinoceptors are involved in the induction of an osmolyte permeability in malaria-infected and oxidized human erythrocytes. FASEB J 20:133–135
Myssina S, Lang PA, Kempe DS, Kaiser S, Huber SM, Wieder T, Lang F (2004) Cl- channel blockers NPPB and niflumic acid blunt Ca(2 +)-induced erythrocyte ‘apoptosis’. Cell Physiol Biochem 14:241–248
Mandal D, Baudin-Creuza V, Bhattacharyya A, Pathak S, Delaunay J, Kundu M, Basu J (2003) Caspase 3-mediated proteolysis of the N-terminal cytoplasmic domain of the human erythroid anion exchanger 1 (band 3). J Biol Chem 278:52551–52558
Matarrese P, Straface E, Pietraforte D, Gambardella L, Vona R, Maccaglia A, Minetti M, Malorni W (2005) Peroxynitrite induces senescence and apoptosis of red blood cells through the activation of aspartyl and cysteinyl proteases. FASEB J 19:416–418
Foller M, Sopjani M, Koka S, Gu S, Mahmud H, Wang K, Floride E, Schleicher E, Schulz E, Munzel T, Lang F (2009) Regulation of erythrocyte survival by AMP-activated protein kinase. FASEB J 23:1072–1080
Foller M, Feil S, Hofmann F, Koka S, Kasinathan R, Nicolay J, Huber S, Feil R, Lang F (2008) Anemia of gene targeted mice lacking functional cGMP-dependent protein kinase type I. Proc Natl Acad Sci USA 105:6771–6778
Kempe DS, Ackermann TF, Fischer SS, Koka S, Boini KM, Mahmud H, Foller M, Rosenblatt KP, Kuro O, Lang F (2009) Accelerated suicidal erythrocyte death in Klotho-deficient mice. Pflugers Arch 458:503–512
Akel A, Wagner CA, Kovacikova J, Kasinathan RS, Kiedaisch V, Koka S, Alper SL, Bernhardt I, Wieder T, Huber SM, Lang F (2007) Enhanced suicidal death of erythrocytes from gene-targeted mice lacking the Cl-/HCO(3)(-) exchanger AE1. Am J Physiol Cell Physiol 292:C1759–C1767
Foller M, Kasinathan RS, Koka S, Huber SM, Schuler B, Vogel J, Gassmann M, Lang F (2007) Enhanced susceptibility to suicidal death of erythrocytes from transgenic mice overexpressing erythropoietin. Am J Physiol Regul Integr Comp Physiol 293:R1127–R1134
Lang PA, Kempe DS, Tanneur V, Eisele K, Klarl BA, Myssina S, Jendrossek V, Ishii S, Shimizu T, Waidmann M, Hessler G, Huber SM, Lang F, Wieder T (2005) Stimulation of erythrocyte ceramide formation by platelet-activating factor. J Cell Sci 118:1233–1243
Foller M, Mahmud H, Koka S, Lang F (2008) Reduced Ca2 + entry and suicidal death of erythrocytes in PDK1 hypomorphic mice. Pflugers Arch 455:939–949
Ghashghaeinia M, Cluitmans JC, Akel A, Dreischer P, Toulany M, Koberle M, Skabytska Y, Saki M, Biedermann T, Duszenko M, Lang F, Wieder T, Bosman GJ (2012) The impact of erythrocyte age on eryptosis. Br J Haematol 157:606–614
Ghashghaeinia M, Cluitmans JC, Toulany M, Saki M, Koberle M, Lang E, Dreischer P, Biedermann T, Duszenko M, Lang F, Bosman GJ, Wieder T (2013) Age sensitivity of NFkappaB abundance and programmed cell death in erythrocytes induced by NFkappaB inhibitors. Cell Physiol Biochem 32:801–813
Lang KS, Myssina S, Brand V, Sandu C, Lang PA, Berchtold S, Huber SM, Lang F, Wieder T (2004) Involvement of ceramide in hyperosmotic shock-induced death of erythrocytes. Cell Death Differ 11:231–243
Lang F, Gulbins E, Lang PA, Zappulla D, Foller M (2010) Ceramide in suicidal death of erythrocytes. Cell Physiol Biochem 26:21–28
Chow SE, Kao CH, Liu YT, Cheng ML, Yang YW, Huang YK, Hsu CC, Wang JS (2014) Resveratrol induced ER expansion and ER caspase-mediated apoptosis in human nasopharyngeal carcinoma cells. Apoptosis 19:527–541
Fonseca BM, Correia-da-Silva G, Teixeira NA (2013) The endocannabinoid anandamide induces apoptosis of rat decidual cells through a mechanism involving ceramide synthesis and p38 MAPK activation. Apoptosis 18:1526–1535
Grassme H, Carpinteiro A, Edwards MJ, Gulbins E, Becker KA (2014) Regulation of the inflammasome by ceramide in cystic fibrosis lungs. Cell Physiol Biochem 34:45–55
Morad SA, Cabot MC (2013) Ceramide-orchestrated signalling in cancer cells. Nat Rev Cancer 13:51–65
Pastukhov O, Schwalm S, Romer I, Zangemeister-Wittke U, Pfeilschifter J, Huwiler A (2014) Ceramide kinase contributes to proliferation but not to prostaglandin E2 formation in renal mesangial cells and fibroblasts. Cell Physiol Biochem 34:119–133
Rossi A, Lord JM (2013) Adiponectin inhibits neutrophil apoptosis via activation of AMP kinase, PKB and ERK 1/2 MAP kinase. Apoptosis 18:1469–1480
Towhid ST, Schmidt EM, Tolios A, Munzer P, Schmid E, Borst O, Gawaz M, Stegmann E, Lang F (2013) Stimulation of platelet death by vancomycin. Cell Physiol Biochem 31:102–112
Barenholz Y, Thompson TE (1980) Sphingomyelins in bilayers and biological membranes. Biochim Biophys Acta 604:129–158
Hakomori S (1983) Chemistry of Glycosphingolipids. Plenum Press, New York and London, pp 1–165
Hannun YA, Obeid LM (2011) Many ceramides. J Biol Chem 286:27855–27862
Oninla VO, Breiden B, Babalola JO, Sandhoff K (2014) Acid sphingomyelinase activity is regulated by membrane lipids and facilitates cholesterol transfer by NPC2. J Lipid Res 55:2606–2619
Schissel SL, Jiang X, Tweedie-Hardman J, Jeong T, Camejo EH, Najib J, Rapp JH, Williams KJ, Tabas I (1998) Secretory sphingomyelinase, a product of the acid sphingomyelinase gene, can hydrolyze atherogenic lipoproteins at neutral pH. Implications for atherosclerotic lesion development. J Biol Chem 273:2738–2746
Hurwitz R, Ferlinz K, Sandhoff K (1994) The tricyclic antidepressant desipramine causes proteolytic degradation of lysosomal sphingomyelinase in human fibroblasts. Biol Chem Hoppe Seyler 375:447–450
Kornhuber J, Tripal P, Reichel M, Terfloth L, Bleich S, Wiltfang J, Gulbins E (2008) Identification of new functional inhibitors of acid sphingomyelinase using a structure-property-activity relation model. J Med Chem 51:219–237
Lang KS, Lang PA, Bauer C, Duranton C, Wieder T, Huber SM, Lang F (2005) Mechanisms of suicidal erythrocyte death. Cell Physiol Biochem 15:195–202
Levy M, Futerman AH (2010) Mammalian ceramide synthases. IUBMB Life 62:347–356
Venkataraman K, Riebeling C, Bodennec J, Riezman H, Allegood JC, Sullards MC, Merrill AH Jr, Futerman AH (2002) Upstream of growth and differentiation factor 1 (uog1), a mammalian homolog of the yeast longevity assurance gene 1 (LAG1), regulates N-stearoyl-sphinganine (C18-(dihydro)ceramide) synthesis in a fumonisin B1-independent manner in mammalian cells. J Biol Chem 277:35642–35649
Laviad EL, Albee L, Pankova-Kholmyansky I, Epstein S, Park H, Merrill AH Jr, Futerman AH (2008) Characterization of ceramide synthase 2: tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J Biol Chem 283:5677–5684
Lahiri S, Futerman AH (2005) LASS5 is a bona fide dihydroceramide synthase that selectively utilizes palmitoyl-CoA as acyl donor. J Biol Chem 280:33735–33738
Mizutani Y, Kihara A, Igarashi Y (2006) LASS3 (longevity assurance homologue 3) is a mainly testis-specific (dihydro)ceramide synthase with relatively broad substrate specificity. Biochem J 398:531–538
Riebeling C, Allegood JC, Wang E, Merrill AH Jr, Futerman AH (2003) Two mammalian longevity assurance gene (LAG1) family members, trh1 and trh4, regulate dihydroceramide synthesis using different fatty acyl-CoA donors. J Biol Chem 278:43452–43459
Mizutani Y, Kihara A, Igarashi Y (2005) Mammalian Lass6 and its related family members regulate synthesis of specific ceramides. Biochem J 390:263–271
Grassme H, Riethmuller J, Gulbins E (2007) Biological aspects of ceramide-enriched membrane domains. Prog Lipid Res 46:161–170
Hannun YA, Obeid LM (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9:139–150
Reinehr R, Haussinger D (2008) CD95 ligation and intracellular membrane flow. Biochem J 413:e11–e12
van Blitterswijk WJ, van der Luit AH, Veldman RJ, Verheij M, Borst J (2003) Ceramide: second messenger or modulator of membrane structure and dynamics? Biochem J 369:199–211
Goni FM, Alonso A (2002) Sphingomyelinases: enzymology and membrane activity. FEBS Lett 531:38–46
Lopez-Montero I, Monroy F, Velez M, Devaux PF (2010) Ceramide: from lateral segregation to mechanical stress. Biochim Biophys Acta 1798:1348–1356
Dinkla S, Wessels K, Verdurmen WP, Tomelleri C, Cluitmans JC, Fransen J, Fuchs B, Schiller J, Joosten I, Brock R, Bosman GJ (2012) Functional consequences of sphingomyelinase-induced changes in erythrocyte membrane structure. Cell Death Dis 3:e410
Brand V, Koka S, Lang C, Jendrossek V, Huber SM, Gulbins E, Lang F (2008) Influence of amitriptyline on eryptosis, parasitemia and survival of Plasmodium berghei-infected mice. Cell Physiol Biochem 22:405–412
Lang KS, Myssina S, Lang PA, Tanneur V, Kempe DS, Mack AF, Huber SM, Wieder T, Lang F, Duranton C (2004) Inhibition of erythrocyte phosphatidylserine exposure by urea and Cl-. Am J Physiol Renal Physiol 286:F1046–F1053
Abed M, Towhid ST, Mia S, Pakladok T, Alesutan I, Borst O, Gawaz M, Gulbins E, Lang F (2012) Sphingomyelinase-induced adhesion of eryptotic erythrocytes to endothelial cells. Am J Physiol Cell Physiol 303:C991–C999
Sheikine Y, Sirsjo A (2008) CXCL16/SR-PSOX–a friend or a foe in atherosclerosis? Atherosclerosis 197:487–495
Ludwig A, Weber C (2007) Transmembrane chemokines: versatile ‘special agents’ in vascular inflammation. Thromb Haemost 97:694–703
Ahmed MS, Langer H, Abed M, Voelkl J, Lang F (2013) The uremic toxin acrolein promotes suicidal erythrocyte death. Kidney Blood Press Res 37:158–167
Nicolay JP, Gatz S, Liebig G, Gulbins E, Lang F (2007) Amyloid induced suicidal erythrocyte death. Cell Physiol Biochem 19:175–184
Zbidah M, Lupescu A, Jilani K, Fajol A, Michael D, Qadri SM, Lang F (2012) Apigenin-induced suicidal erythrocyte death. J Agric Food Chem 60:533–538
Malik A, Bissinger R, Calabro S, Faggio C, Jilani K, Lang F (2014) Aristolochic acid induced suicidal erythrocyte death. Kidney Blood Press Res 39:408–419
Biswas D, Banerjee M, Sen G, Das JK, Banerjee A, Sau TJ, Pandit S, Giri AK, Biswas T (2008) Mechanism of erythrocyte death in human population exposed to arsenic through drinking water. Toxicol Appl Pharmacol 230:57–66
Mahmud H, Foller M, Lang F (2009) Arsenic-induced suicidal erythrocyte death. Arch Toxicol 83:107–113
Bissinger R, Malik A, Honisch S, Warsi J, Jilani K, Lang F (2014) In vitro sensitization of erythrocytes to programmed cell death following baicalein treatment. Toxins (Basel) 6:2771–2786
Lang E, Jilani K, Zelenak C, Pasham V, Bobbala D, Qadri SM, Lang F (2011) Stimulation of suicidal erythrocyte death by benzethonium. Cell Physiol Biochem 28:347–354
Braun M, Foller M, Gulbins E, Lang F (2009) Eryptosis triggered by bismuth. Biometals 22:453–460
Bentzen PJ, Lang E, Lang F (2007) Curcumin induced suicidal erythrocyte death. Cell Physiol Biochem 19:153–164
Bobbala D, Koka S, Lang C, Boini KM, Huber SM, Lang F (2008) Effect of cyclosporine on parasitemia and survival of Plasmodium berghei infected mice. Biochem BiophysRes Commun 376:494–498
Abed M, Zoubi KA, Theurer M, Lang F (2013) Effect of dermaseptin on erythrocytes. Basic Clin Pharmacol Toxicol 113:347–352
Calabro S, Alzoubi K, Bissinger R, Faggio C, Lang F (2014) Stimulation of suicidal erythrocyte death by ellipticine. Basic Clin Pharmacol Toxicol. doi:10.1111/bcpt.12350; [Epub ahead of print]
Bissinger R, Modicano P, Frauenfeld L, Lang E, Jacobi J, Faggio C, Lang F (2013) Estramustine-induced suicidal erythrocyte death. Cell Physiol Biochem 32:1426–1436
Jilani K, Enkel S, Bissinger R, Almilaji A, Abed M, Lang F (2013) Fluoxetine induced suicidal erythrocyte death. Toxins (Basel) 5:1230–1243
Eberhard M, Ferlinz K, Alizzi K, Cacciato PM, Faggio C, Foller M, Lang F (2010) FTY720-induced suicidal erythrocyte death. Cell Physiol Biochem 26:761–766
Zbidah M, Lupescu A, Jilani K, Lang F (2013) Stimulation of suicidal erythrocyte death by fumagillin. Basic Clin Pharmacol Toxicol 112:346–351
Lupescu A, Jilani K, Zelenak C, Zbidah M, Shaik N, Lang F (2012) Induction of programmed erythrocyte death by gambogic acid. Cell Physiol Biochem 30:428–438
Jilani K, Qadri SM, Lang F (2013) Geldanamycin-induced phosphatidylserine translocation in the erythrocyte membrane. Cell Physiol Biochem 32:1600–1609
Gatidis S, Foller M, Lang F (2009) Hemin-induced suicidal erythrocyte death. Ann Hematol 88:721–726
Lupescu A, Jilani K, Zelenak C, Zbidah M, Qadri SM, Lang F (2012) Hexavalent chromium-induced erythrocyte membrane phospholipid asymmetry. Biometals 25:309–318
Zbidah M, Lupescu A, Herrmann T, Yang W, Foller M, Jilani K, Lang F (2013) Effect of honokiol on erythrocytes. Toxicol In Vitro 27:1737–1745
Ahmed MS, Abed M, Voelkl J, Lang F (2013) Triggering of suicidal erythrocyte death by uremic toxin indoxyl sulfate. BMC Nephrol 14:244
Calabro S, Alzoubi K, Bissinger R, Jilani K, Faggio C, Lang F (2014) Enhanced eryptosis following juglone exposure. Basic Clin Pharmacol Toxicol. doi:10.1111/bcpt.12340; [Epub ahead of print]
Bhavsar SK, Bobbala D, Xuan NT, Foller M, Lang F (2010) Stimulation of suicidal erythrocyte death by alpha-lipoic acid. Cell Physiol Biochem 26:859–868
Alzoubi K, Alktifan B, Oswald G, Fezai M, Abed M, Lang F (2014) Breakdown of phosphatidylserine asymmetry following treatment of erythrocytes with lumefantrine. Toxins (Basel) 6:650–664
Eisele K, Lang PA, Kempe DS, Klarl BA, Niemoller O, Wieder T, Huber SM, Duranton C, Lang F (2006) Stimulation of erythrocyte phosphatidylserine exposure by mercury ions. Toxicol Appl Pharmacol 210:116–122
Mahmud H, Foller M, Lang F (2008) Stimulation of erythrocyte cell membrane scrambling by methyldopa. Kidney Blood Press Res 31:299–306
Arnold M, Bissinger R, Lang F (2014) Mitoxantrone-induced suicidal erythrocyte death. Cell Physiol Biochem 34:1756–1767
Arnold M, Lang E, Modicano P, Bissinger R, Faggio C, Abed M, Lang F (2014) Effect of nitazoxanide on erythrocytes. Basic Clin Pharmacol Toxicol 114:421–426
Jilani K, Lupescu A, Zbidah M, Abed M, Shaik N, Lang F (2012) Enhanced apoptotic death of erythrocytes induced by the mycotoxin ochratoxin A. Kidney Blood Press Res 36:107–118
Jilani K, Qadri SM, Zelenak C, Lang F (2011) Stimulation of suicidal erythrocyte death by oridonin. Arch Biochem Biophys 511:14–20
Koka S, Bobbala D, Lang C, Boini KM, Huber SM, Lang F (2009) Influence of paclitaxel on parasitemia and survival of Plasmodium berghei infected mice. Cell Physiol Biochem 23:191–198
Lang PA, Huober J, Bachmann C, Kempe DS, Sobiesiak M, Akel A, Niemoeller OM, Dreischer P, Eisele K, Klarl BA, Gulbins E, Lang F, Wieder T (2006) Stimulation of erythrocyte phosphatidylserine exposure by paclitaxel. Cell Physiol Biochem 18:151–164
Alzoubi K, Honisch S, Abed M, Lang F (2014) Triggering of suicidal erythrocyte death by penta-O-galloyl-beta-d-glucose. Toxins (Basel) 6:54–65
Foller M, Biswas R, Mahmud H, Akel A, Shumilina E, Wieder T, Goetz F, Lang F (2009) Effect of peptidoglycans on erythrocyte survival. Int J Med Microbiol 299:75–85
Bissinger R, Malik A, Warsi J, Jilani K, Lang F (2014) Piperlongumine-induced phosphatidylserine translocation in the erythrocyte membrane. Toxins (Basel) 6:2975–2988
Lupescu A, Jilani K, Zbidah M, Lang E, Lang F (2012) Enhanced Ca2 + entry, ceramide formation, and apoptotic death of erythrocytes triggered by plumbagin. J Nat Prod 75:1956–1961
Abed M, Towhid ST, Shaik N, Lang F (2012) Stimulation of suicidal death of erythrocytes by rifampicin. Toxicology 302:123–128
Lupescu A, Jilani K, Zbidah M, Lang F (2012) Induction of apoptotic erythrocyte death by rotenone. Toxicology 300:132–137
Bissinger R, Modicano P, Alzoubi K, Honisch S, Faggio C, Abed M, Lang F (2014) Effect of saponin on erythrocytes. Int J Hematol 100:51–59
Sopjani M, Foller M, Gulbins E, Lang F (2008) Suicidal death of erythrocytes due to selenium-compounds. Cell Physiol Biochem 22:387–394
Lupescu A, Bissinger R, Jilani K, Lang F (2014) In vitro induction of erythrocyte phosphatidylserine translocation by the natural naphthoquinone shikonin. Toxins (Basel) 6:1559–1574
Lupescu A, Shaik N, Jilani K, Zelenak C, Lang E, Pasham V, Zbidah M, Plate A, Bitzer M, Foller M, Qadri SM, Lang F (2012) Enhanced erythrocyte membrane exposure of phosphatidylserine following sorafenib treatment: an in vivo and in vitro study. Cell Physiol Biochem 30:876–888
Qadri SM, Bauer J, Zelenak C, Mahmud H, Kucherenko Y, Lee SH, Ferlinz K, Lang F (2011) Sphingosine but not sphingosine-1-phosphate stimulates suicidal erythrocyte death. Cell Physiol Biochem 28:339–346
Alzoubi K, Calabro S, Faggio C, Lang F (2014) Stimulation of Suicidal Erythrocyte Death by Sulforaphane. Basic Clin Pharmacol Toxicol
Zbidah M, Lupescu A, Yang W, Bosc A, Jilani K, Shaik N, Lang F (2012) Sulindac sulfide–induced stimulation of eryptosis. Cell Physiol Biochem 30:1072–1082
Shaik N, Lupescu A, Lang F (2012) Sunitinib-sensitive suicidal erythrocyte death. Cell Physiol Biochem 30:512–522
Abed M, Herrmann T, Alzoubi K, Pakladok T, Lang F (2013) Tannic acid induced suicidal erythrocyte death. Cell Physiol Biochem 32:1106–1116
Zelenak C, Pasham V, Jilani K, Tripodi PM, Rosaclerio L, Pathare G, Lupescu A, Faggio C, Qadri SM, Lang F (2012) Tanshinone IIA stimulates erythrocyte phosphatidylserine exposure. Cell Physiol Biochem 30:282–294
Nguyen TT, Foller M, Lang F (2009) Tin triggers suicidal death of erythrocytes. J Appl Toxicol 29:79–83
Frauenfeld L, Alzoubi K, Abed M, Lang F (2014) Stimulation of erythrocyte cell membrane scrambling by mushroom tyrosinase. Toxins (Basel) 6:1096–1108
Jilani K, Abed M, Zelenak C, Lang E, Qadri SM, Lang F (2011) Triggering of erythrocyte cell membrane scrambling by ursolic acid. J Nat Prod 74:2181–2186
Qadri SM, Eberhard M, Mahmud H, Föller M, Lang F (2009) Stimulation of ceramide formation and suicidal erythrocyte death by vitamin K(3) (menadione). Eur J Pharmacol 623:10–13
Jilani K, Lupescu A, Zbidah M, Shaik N, Lang F (2013) Withaferin A-stimulated Ca2 + entry, ceramide formation and suicidal death of erythrocytes. Toxicol In Vitro 27:52–58
Kiedaisch V, Akel A, Niemoeller OM, Wieder T, Lang F (2008) Zinc-induced suicidal erythrocyte death. Am J Clin Nutr 87:1530–1534
Acknowledgments
The authors acknowledge the meticulous preparation of the manuscript by Tanja Loch and Lejla Subasic. Their research has been supported by the Deutsche Forschungsgemeinschaft, Nr. La 315/4-3, La 315/6-1, La 315/13-1.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lang, E., Bissinger, R., Gulbins, E. et al. Ceramide in the regulation of eryptosis, the suicidal erythrocyte death. Apoptosis 20, 758–767 (2015). https://doi.org/10.1007/s10495-015-1094-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-015-1094-4