
Abstract
Innate immunity, which is the first line of host defense against invading microbial pathogens in multicellular organisms, occurs through germline-encoded pattern-recognition receptors. The Toll-like receptor/Interleukin (IL)-1 receptor (TLR/IL-1R) superfamily comprises proteins that contain the phylogenetically conserved Toll/IL-1 receptor (TIR) domain, which is responsible for the propagation of downstream signaling through recruitment of TIR domain containing cytosolic adaptor proteins such as MyD88, TIRAP/MAL, TRIF, TRAM and SARM. These interactions activate transcription factors that regulate the expression of various proinflammatory cytokines (IL-1, IL-6, IL-8 and TNF-α) and chemokines. Activation of the TLR/IL-1R signaling pathway promotes the onset of inflammatory diseases, autoimmune diseases and cancer; therefore, this pathway can be used for the development of therapeutic strategies against these types of pathogenesis. In this review paper, we illustrate the role of the TIR–TIR domain interaction with the TLR/IL-1R signaling pathway in inflammation and apoptosis and recent therapeutic drugs targeted to inhibit the downstream signaling cascade for treatment of inflammatory diseases and cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Every living organism fights against invading microorganisms to protect itself from infection through innate and acquired immunity. In vertebrates, acquired immunity responses are slower process in which T cells and B cells express highly diverse antigen receptors generated through DNA rearrangements of different variable (V) region gene segments with diversity (D) and joining (J) gene segments to respond to a wide range of potential antigens, whereas the innate immune system constitutes the first line of host defense against invading microbial pathogens and other endogenous danger signals. Innate immunity is phylogenetically conserved and present in almost all multicellular organisms [1]. In innate immunity, pathogen-associated or danger-associated molecular patterns (PAMPs/DAMPs) are recognized by pattern-recognition receptors (PRRs) such as membrane-bound Toll-like receptors (TLRs) and C-type lectin receptors (CLRs), or cytosolic nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) and retinoic acid-inducible gene (RIG)-I-like receptors (RLRs) [2–4]. After PAMPs/DAMPs recognition, these PRR receptors in various cell compartments recruit specific adaptor proteins that determine the specificity of inflammatory response via activation of distinct transcription factors and pro-inflammatory genes [5]. This innate immunity has a greater degree of ability to discriminate between self and foreign pathogens [6]. The activation of innate immunity is essential to the induction of acquired immunity, especially in the induction of T helper 1 (TH1)-cell response [7]. The Toll/interleukin-1 receptor (TIR) family, which comprises TLRs and interleukin-1 receptors (IL-1Rs), is prerequisite for many host innate immune responses. Signal transduction through these receptors leads to the activation of transcription factors NF-κB and activator protein 1 (AP-1). In recent years, knowledge regarding TIR domain signaling and its responses has increased, which has improved our understanding of the pathogenesis and treatment of cancers, pathogenic infections and immune and allergic diseases [8]. In this review, we discuss the TIR signaling pathways and the role of the TIR domain in inflammation and apoptosis.
Toll/interleukin-1 receptor (TIR) superfamily
The members of TLRs and interleukin (IL)-1 type I receptors constitute the TLR/IL-1R superfamily, which plays a fundamental role in immune response. Toll/IL-1 receptor (TIR) domain is a cytoplasmic conserved region present in TLRs and IL-1R. The induction of cyclooxygenase type 2 (COX-2), increased expression of adhesion molecules, tissue degrading enzymes, chemokines, or synthesis of nitric oxide (NO) are inflammatory responses to both TLR and IL-1R ligands. Upon binding of ligands to TLRs or IL-1Rs, adaptor proteins such as MyD88, MAL/TIRAP, TRIF, and TRAM are recruited to the cytoplasmic region of TIR domains of the receptors through TIR–TIR interaction.
Toll-like receptors (TLRs) subfamily
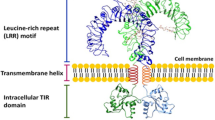
Toll-like receptors (TLRs) are evolutionarily conserved receptors of type I integral transmembrane glycoproteins with trimodular structure that respond to a wide variety of endogenous ligands (HSP60, HSP70, endoplasmin, HSPB8, α-crystallin A chain, HMGB1, uric acid crystals, surfactant protein A, fibronectin, heparan sulfate, biglycan, fibrinogen, oligosaccharides of hyaluronan) and exogenous ligands such as microbial pathogens (bacteria, mycobacteria, mycoplasma, fungi, protozoa and virus) that serve as important components of the innate immune system [9]. In Drosophila melanogaster embryogenesis, Toll protein helps establish dorsoventral polarity. In 1996, Toll protein was found to play a role in adult Drosophila immunity against fungal infection [10]. Moreover, Drosophila TLR, 18-Wheeler has been implicated in bacterial infections [11]. In 1997, Janeway et al. identified the first human TLR, and 13 TLRs have been identified in mammals to date. TLRs function as PPRs recognizing PAMPs such as lipopolysaccharides, lipoproteins, and nucleic acids [12]. Although TLR1-TLR9 are conserved between humans and mice, TLR10 is not functional in mice because of a retrovirus insertion, and TLR11, TLR12 and TLR13 are lost in human genomes [13].
TLRs are composed of three major domains, (i) the extracellular N-terminal domain (ectodomain), which recognizes their respective PAMPs, consists of approximately 19–25 leucine-rich repeats (LRRs) that each contain 24–29 amino acids with the conserved motif “XLXXLXLXX” folded in β-strands and in α-helices that are linked by loops, (ii) a transmembrane domain, and (iii) an intracellular C-terminal domain known as the Toll/IL-1 receptor (TIR) domain [14]. The ectodomain forms a horseshoe structure with a concave surface that is involved in the recognition of various pathogens. The TIR domain is required for the interaction and recruitment of various adaptor molecules to activate the downstream signaling pathway [15]. TLRs are expressed on various hematopoietic cells including macrophages, dendritic cells, B cells and T cells [16, 17], as well as on a variety of non-hematopoietic cells including epithelial cells, endothelial cells and fibroblasts [18, 19]. TLRs are expressed in distinct cellular compartments such as the cell surface, endosomes, lysosomes, or cytoplasm. Human TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10 and mouse TLR11 and TLR12 are expressed largely on the cell surface and recognize microbial membrane components such as lipoproteins, lipids and proteins [13, 20], whereas TLR3, TLR7, TLR8, and TLR9 are localized in intracellular vesicles such as the endosome or lysosome and endoplasmic reticulum (ER) and recognize microbial nucleic acid species. The intracellular TLRs are expressed on the ER in resting cells and trafficked to the endosomal compartment in response to PAMP stimulation. UNC93B, 12-membrane-spanning ER proteins, interacts with transmembrane regions of TLR3, TLR7 and TLR9 in the ER and assists in the trafficking of TLR7 and TLR9 from the ER to the endosome [21]. TLR1 (which is also identified as TIL) is ubiquitously highly expressed than other TLRs. TLR2 (TIL4) is expressed in lymphoid tissue, monocytes and peripheral blood lymphocytes. TLR3 is expressed in the heart, brain, lung and muscle. TLR4 expression predominates the spleen and lymphocytes, as well as the heart. TLR5 (TIL3) is expressed in the prostrate, ovary, peripheral blood monocytes and leukocytes. TLR6, which is closely related in sequence to TLR1, is predominantly expressed in the spleen, thymus, lung and ovary [22] (Table 1).
Interleukin-1 receptor (IL-1R) subfamily
The interleukin-1 receptor (IL-1R) subfamily plays a central role in regulation of innate inflammatory and immune responses to infections, injury, stress, and allergies [23] (Fig. 1). Gay and Keith [24] found that the cytoplasmic domain of IL-1R is homologous with the cytoplasmic TIR domain of Toll protein of Drosophila, which makes the biochemical nature of signal transduction similar in both cases. Activation of IL-1R by IL-1 causes nuclear localization of the transcriptional activator, NF-κB. The inactive cytoplasmic form of NF-κB is complexed with Iκβ, and the phosphorylation of Iκβ by protein kinase C causes it to dissociate, enabling NF-κB to migrate to the nucleus to regulate immune and inflammatory responses. The IL-1R type I family encodes ten members with three domains. The extracellular domain displays homology to immunoglobulin-like (IgG), a transmembrane domain and a cytoplasmic domain of the IL-1 type I receptor, which is highly homologous with the cytoplasmic domain of all TLRs [25]. The Ig-like domains of the IL-IR family members display an Ig fold, which consists of two β-pleated sheets held together by intradomain disulfide bonds via conserved cysteine residues. Extracellular Ig-like domain is involved in protein–ligand and protein–protein interactions [26]. In humans, the three extracellular Ig domains of the IL-1RI family have six amino acids, Arg431, Lys515, Arg518, Phe513, Trp514, and Tyr519, which are essential to signaling. In addition, Pro521 is required for the maximum signaling capacity, and Phe513 and Trp514 are present in the conserved box 3 of the TIR domain of IL-1RI [27]. IL-1R1, IL-1R2, and IL-1 receptor accessory protein (IL-1RAcP)/(IL-1R3) are bona fide receptors for IL-1α and IL-1β [28]. ST2, which is also known as IL-R4, contains ligand IL-33. IL-R5, which is the ligand-binding (α) chain of the IL-18 receptor, is termed as IL-18Rα. Interleukin-1 family (IL-1F) ligands such as IL-1F6, IL-1F8, and IL-1F9 are pro-inflammatory cytokines that bind to IL-1R-related protein (IL-1Rrp2) as its ligand-binding (α) chain and recruit IL-1RAcP as a co-receptor (β) chain to activate NF-κB. IL-1R7, which is also known as IL-18Rβ, is a co-receptor (β) chain involved in IL-18 signal transduction. IL-1R8 and IL-1R9 are encoded in X chromosomes and expressed in fetal brains. Both of these are homologous to IL-1RAcP and IL-18Rβ. IL-1R8 and IL-1R9 are homologous and referred to as three Ig IL-1 related receptors (TIGIRR). Ligands for IL-1R8 and IL-1R9 are not known. Single Ig IL-1 related receptor (SIGIRR) contains only one IgG domain for the extracellular segment and possesses the longest cytoplasmic domain of all members of the IL-1 receptor family. This receptor is a negative regulator of both IL-1α and IL-1β activities that functions as an anti-inflammatory receptor suppressing inflammation [25, 29] (Table 2).

Interleukin-1 (IL-1) family of receptors
TIR-domain adaptor proteins
Signaling by TLRs and IL-1Rs family members involves adaptor proteins such as MyD88, TIRAP/MAL, TRIF, TRAM and SARM. These adaptors interact to activate transcription factors such as NF-κB, IRF1, IRF3, IRF5, and IRF7 and interferon-γ-signaling.
MyD88
Myeloid differentiation primary-response protein 88 (MyD88) has an amino (N)-terminal death domain (DD), followed by a shorter linker sequence and a carboxy (C)-terminal TIR domain. It also has an intermediate domain (ID), which is essential to TLR signaling due to its interaction with IL-1R-associated kinases 4 (IRAK4) [30]. MyD88 has been shown to be induced during IL-6-stimulated differentiation of M1 myeloid leukemia cells into macrophages [31]. During IL-1R activation by IL-1 cytokine, MyD88-TIR interacts with TIR domain of IL-1R1 complex through homophilic interaction and recruits IRAK through DD interaction. The knockout of MyD88 in mice showed no responses to TLR4 ligand LPS, TLR2 ligand peptidoglycan and lipoproteins, TLR9 ligand unmethylated CpG DNA, TLR7 ligand imidazoquinoline or TLR5 ligand flagellin. Taken together, these findings demonstrated the role of MyD88 in inflammatory responses mediated by TLR family members [32–36].
TIRAP/MAL
Toll/interleukin 1 receptor (TIR) domain containing adaptor protein (TIRAP)/MyD88 adaptor-like (MAL) protein possesses a TIR domain in the cytoplasmic tail (C-terminus) and belongs to the TLR/IL-1R superfamily. However, MAL does not contain DD domain [37]. The interaction of TIR domain in the adaptor protein is essential to downstream signaling. MAL acts as a bridging adaptor protein between TLR1/2, TLR2/6 and TLR4 and MyD88. Thus, MAL recruits MyD88 and is critically involved in the MyD88-dependent signaling pathway through TLR2 and TLR4, but is not associated with MyD88-independent signaling [38, 39].
TRIF
Another adaptor molecule is TIR-domain-containing adaptor protein inducing IFN-β (TRIF), which is also known as TIR-domain-containing molecule 1 (TICAM1). TRIF is a large protein containing 712 amino acids in humans. Overexpression of TRIF along with MyD88 and MAL activated NF-κB-dependent promoters in human embryonic kidney 293 (HEK-293) cells, whereas overexpression of TRIF alone induced activation of interferon-β (IFN-β) promoter. TRIF knockout mice were defective in both TLR3- and TLR4-mediated expression of IFN-β production and IFN-inducible genes [40]. However, TLR4-mediated activation of the MyD88-dependent pathway through phosphorylation of IRAK1 and early-phase activation of NF-κB was not impaired. Thus, TRIF is essential to induction of the expression of inflammatory cytokines facilitating mammalian antiviral host defense via TLR3- and TLR4-mediated MyD88-independent pathways [41]. Overall, TRIF associates with TLR3 and TLR4, binding directly to TLR3 and using another adaptor protein, TRAM, to bind to TLR4.
TRAM
This molecule is known as TRIF-related adaptor molecule (TRAM) or TIR-domain-containing molecule 2 (TICAM2). TRAM was the fourth TIR domain-containing adaptor identified through sequence homology in database searches [42, 43]. TRAM knockout mice showed impaired activation of IRF3 and reduced expression of IFN-inducible genes in response to TLR4 stimulation. However, unlike TRIF-deficient mice, TRAM-deficient mice showed a response to TLR3 stimulation [44, 45]. Based on these findings, TRAM clearly associates with TRIF and TLR4, but not with TLR3, and is specifically involved in activation of the MyD88-independnet/TRIF-dependent signaling pathway through TLR4.
SARM
Sterile α- and HEAT/Armadillo motif containing protein (SARM) is another highly conserved TIR domain-containing adaptor protein identified through searches of the human genome in 2001 [46]. SARM contains 690 amino acids with a high degree of sequence similarity to proteins in Drosophila melanogaster and Caenorhabditis elegans. This protein contains a TIR domain at the C-terminus, two sterile α motif (SAM) domains and an Armadillo repeat motif (ARM), which is annotated as SARM. SAM domain is present in nuclear proteins and involved in the development by homo- and hetero-oligomerization facilitating protein–protein interactions. ARM is a 40 amino acid tandem repeat that mediates the interaction of β-catenin with its ligands [47]. In vitro studies showed that human SARM inhibits the function of adaptor protein TRIF, which mediates the MyD88-independent signaling of TLR3 and TLR4, blocking the induction of proinflammatory genes [48, 49]. Thus, SARM negatively regulates TRIF downstream signaling.
Structure of TIR domains
The TIR domain of TLRs and IL-1Rs show a conserved cytoplasmic region of 135–160 amino acids (Fig. 2). Within the TIR domain of TLRs, there are three conserved boxes in the region of homology that are crucial to signaling. Box 1 and 2 are involved in binding downstream signaling molecules, while box 3 is involved in direct localization of the TLR/IL-1 receptor through interactions with cytoskeletal elements [26, 50]. TIR domain facilitates homotypic protein–protein interactions during signal transduction. Details on the structure and function of TIR domains are exclusively described in this special issue by Kobe and colleagues. The crystal structure of the TIR domain of human TLR1, TLR2 and TLR10 showed that it contains a central five-stranded parallel β-sheet (βA- βE) surrounded by a total of five α-helices (αA-αE) on both sides that are connected by eight loops (Fig. 2). The residues in β-strands or α-helices are numbered based on their position in strand or helix. The loops are named by the letters of the secondary structure elements that they connect. For example, BB loop connects the βB strand and αB helix [51, 52]. The crystal structures reveal that the core TIR domain of TLR1 and TLR2 starts from the conserved (F/Y)DA amino acid motif and ends roughly eight residues carboxy-terminal to the conserved FW motif. Most of the conserved amino acids are present in the hydrophobic core of the structure, and the surface exposed hydrophilic amino acids vary greatly between TIR domains. Although there is more than 50 % similarity in amino acid sequence, TIR domains of TLR1 and TLR2 differ conformationally, especially in helices αB, αC’, and αD. Although there is 20–30 % sequence conservation among TIR domains, the sizes of the domains vary with the amount of sequence and structural diversity. This TLR structural diversity facilitates specificity, ensuring formation of a proper signaling complex in signal transduction. In TLRs signaling, three types of TIR domain interactions are possible. 1. The “R face” interface mediates oligomerization of receptor TIR domains, which is facilitated by the ligand-induced association of the ecto-domains of the receptors. The interaction that occurs at the R face is the main determinant of specificity in the TLR signaling process in addition to receptor-ligand compatibility. 2. The “A face” interface mediates oligomerization of the TIR domains of the downstream adaptor molecule, which may be facilitated by death domain (DD) interactions in this molecule [53]. 3. The “S face” interface mediates the association between the receptor and adapter TIR domains, and the formation of this TIR domain complex is essential to TLR signaling [54]. Khan et al. [55] reported the first crystal structure of a TIR domain of the IL-1R superfamily, human IL-1R accessory protein-like (IL-1RAPL). There are large structural differences between the TIR domain of IL-1RAPL and that of human TLR1 and TLR2. For example, the structure of the TIR domain of IL-1RAPL contains a central five-stranded fully parallel β-sheet surrounded by helices on both sides. The IL-1RAPL backbone fold is the same as that of the TIR domain of human TLR1 and TLR2. For the β-sheet, the βE strand of IL-1RAPL is longer at the N-terminal end than the βE strand of TLR1. On one face of the β-sheet, the axis of the αD helix in IL-1RAPL is oriented almost perpendicular to that of the αD helix in TLR1 and TLR2. On the other face of the β-sheet, the beginning of the αA helix has different conformations in IL-1RAPL and TLR1 because IL-1RAPL has an insertion between βA and αA. There is also difference in the CD and EE loops between the two TIR domains of IL-1RAPL and TLR1. The BB loop of IL-1RAPL contains a unique hydrogen bond between Thr residues at the 8th and 10th positions. In the TIR domain of IL-1RAPL, there are two short helices followed by a third, well ordered helix (αC’), whereas in the TIR domain of TLR1 and TLR2, there are two short helices (αC and αC’) followed by a highly disordered region. The crystal structure of adaptor molecule MyD88 TIR domain was also solved [56]. The TIR domain of MyD88 comprises a central five-stranded parallel β-sheet (βA- βE) surrounded by four α-helices (αA–αC and αE). The TIR domain of MyD88 is different from other TLRs-TIR domains as it contains five central β-strands surrounded by four α-helices instead of five α-helices. Of all known structures of the TIR domain, the MyD88 TIR domain shows the highest sequence similarity to TLR2. The largest structural discrepancy in the MyD88 TIR domain was observed in the region from the BB loop (Ser194 to Ala208) to αB relative to TLR2. The BB loop was exposed to solvent in both MyD88 and TLR2, but the direction of the loop orientation differed. This was mainly due to structural differences in the C-terminal region of the BB loop, which precedes αB of MyD88; therefore, αB of MyD88 was much shorter than TLR2. There was no α-helix in the region between the βD and βE strands (residues 257–273) of the MyD88 TIR domain. The crystal structure of the MyD88-adapter like (MAL)/TIR-domain containing an adapter protein (TIRAP) was reported [57]. MAL-TIR showed 24 % sequence similarity with MyD88-TIR and 23 % with TLR2-TIR. MAL-TIR possesses an asymmetric unit composed of a TIR domain fold containing a five stranded parallel β-sheet (βA-βE) surrounded by four α-helices (αA and αC-αE). However, it lacks a helical αB segment between the βB- and βC-strands and contains an AB long loop connecting the αA helix and the βB strand. The high-resolution structure of the TLR6 TIR domain comprises a five-stranded or a four-stranded parallel β-sheet surrounded by four or five α-helices. The structure also contains a well-conserved BB loop and DD loop. In the TLR6 structure, the BB loop is not essential to the homo-dimeric interaction, whereas αC-including Cys712 is primarily involved in the interaction [58]. The crystal structure of the TLR5 ectodomain and TIR in the absence of flagellin was reported [59]. The MAL-TIR domain structure revealed an extraordinarily long AB loop, but no αB helix or BB loop. The AB loop is capable of mediating direct binding to the TIR domains of TLR4 and MyD88 simultaneously [60] (Fig. 2).

Structures of TLR/IL-1R superfamily TIR domains
TLR/IL-1R signaling pathways
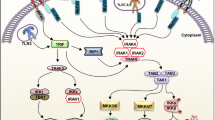
Upon recognition of cognate ligands, TLR/IL-1R either homo- or hetero dimerizes to form TLR1/2, TLR2/6, and IL-1R/IL-1RacP, which activate the downstream signaling cascade through recruitment of the TIR-domain containing adaptor proteins to their TIR domain of the cytoplasmic region (Fig. 3). MyD88 is an essential adaptor protein for all TLR/IL-1R superfamily downstream signaling except for TLR3. MyD88 interacts directly with TLR7, TLR8 and TLR9 through its C-terminal TIR domain and associates with all other TLRs via the adaptor protein TIRAP/MAL. However, following activation of the TLR/IL-1 receptors, the downstream signaling pathway follows the MyD88-dependent pathway and/or MyD88-independent pathway/TRIF-dependent pathway.

TLR/IL-1R inflammatory and apoptotic signaling pathways
In the MyD88-dependent pathway, upon stimulation of TLR/IL-1R receptors, MyD88 is recruited to its respective TIR domains through homotypic interactions. This further allows MyD88 to interact with IRAK family members (IRAK1, IRAK2 and IRAK4) through interaction of its death domain (DD) with the respective DDs present in the amino-terminal region of IRAKs. In particular, the residues located in both the DD and ID domain of MyD88 are mainly involved in the interaction with IRAK-4 [61]. MyD88 ultimately forms macromolecular complexes with IRAK4, IRAK1 and/or IRAK2. IRAK4 phosphorylates IRAK1 and IRAK2 and promotes its association with a RING domain E3 ubiquitin ligase, TRAF6. Activated TRAF6 undergoes K63-polyubiquitination and acts as a platform for binding of TAB 1, TAB 2 and TAB 3. TAK1 ubiquitin-dependent kinase is activated by recruitment to TRAF6/TAB 1/TAB 2/TAB 3. Thus, TRAF6 in conjunction with a dimeric Ub-conjugating enzyme complex consisting of Ubc13 and Uev1A/Mms2 catalyzes formation of a polyubiquitination chain linked through lysine(K)-63 of ubiquitin to recruit TAK1 through the TAB 2 and TAB 3 ubiquitin binding proteins [62, 63]. TAB 2 and TAB 3 bind to lysine 63-linked polyubiquitin chains through a highly conserved zinc finger (ZnF) domain [63]. Activated TAK1 phosphorylates and activates IKKβ (of complex IKK), which further phosphorylates IκBα for its K48-polyubiquitination and subsequent proteasomal degradation, rendering the release of NF-κB (p50/p65) through the canonical pathway, which translocates into the nucleus from cytosol to activate NF-κB-dependent genes [20, 64]. Activated TAK1 activates JNK through MKK4/7, p38 MAPK through MKK3/6 and ERK1/2 through MEK1/2. Thus, the sequential phosphorylation of MAPKKK, MAPKK and MAPK activates activating protein-1 (AP-1) transcription factor [65, 66]. AP-1 is a family of pleiotropic dimeric transcription factors composed of Jun, Fos or ATF (activating transcription factor) subunits that bind TPA-response elements or cAMP-response elements and are involved in cellular proliferation, transformation and death [67, 68]. However, the concomitant activation of transcription factors NF-κB and AP-1 induces a pleiotropic inflammatory response through the production of proinflammatory cytokines including IL-12, IL-6 and TNF-α [32, 69]. The activation of NF-κB and MAPKs is tightly regulated by ubiquitination and phosphorylation, which is responsible for the production of pro-inflammatory cytokines. MAPKs also play a critical role in the regulation of several cellular processes, including proliferation, differentiation and apoptosis [70]. In plasmacytoid dendritic cells (pDCs), TLR7/8 and TLR9 ligands induce a signaling complex, MyD88-IRAK4-TRAF6, which recruits TRAF3, IRAK1, IKKα, osteopontin (OPN) and IRF7. IRF7 is phosphorylated by IRAK1 and IKKα, which forms a dimer and translocates into the nucleus to express IFNα and IFNβ genes.
In the MyD88-independent/TRIF-dependent pathway, stimulation of TLR3 and TLR4 by LPS, poly IC and viral infection all activate transcription factor interferon regulatory factor 3 (IRF3), even in MyD88-deficient cells [70]. The binding of adaptor protein TRIF to the TLR3/4-TIR domain activates dimerization of IRF3 by phosphorylation via non-canonical IκB kinases (IKK), TANK-binding kinase 1 (TBK1) and IKKi/IKKε. TBK1/IKKi is activted via TRAF3-mediated Lys(K)-63-linked ubiquitination and acts as a linking protein between TRIF and TBK1/IKKi [71]. Thus, activated dimerized IRF3 translocates into the nuclei and induces type I IFN, particularly IFNβ. However, secreted IFNβ activates neighboring cells and the JAK-STAT pathway through type I IFN receptor to induce IRF7 expression [72–74]. The TRIF-dependent pathway always activates NF-κB and MAP kinases via two different signaling pathways; specifically, TRAF6 interacts through the N-terminus of TRIF to activate NF-κB and MAP kinases. The C-terminus of TRIF harboring the receptor-interacting protein (RIP) homotypic interaction motif (RHIM) domain interacts with RIP1 and RIP3. Two additional proteins, TNF receptor-associated death domain (TRADD) and Fas-associated death domain protein (FAD), form a complex with RIP1, which then activates NF-κB [75] (Fig. 3).
TIR domain in inflammation
Inflammation is a response to cell/tissue damage by pathogens, physical injury, or noxious stimuli from chemicals. Acute inflammation is a short-term response to repair tissue or enable healing of a damaged region by infiltrated leukocytes, whereas chronic inflammation is a prolonged, dysregulated and maladaptive response that involves active inflammation and impaired tissue repair/tissue destruction. Controlled inflammatory responses are necessary for host defense; however, uncontrolled inflammation results in inflammatory diseases, autoimmune diseases and cancer. Molecular events triggered by the TLR/IL-1R superfamily are initiated by downstream signaling transducers, MyD88 and TRIF, as well as other signaling components, which ultimately leads to activation of NF-κB and initiates the innate inflammatory responses [76]. NF-κB is a major inflammatory switch that comprises a family of transcription factors that regulate expression of various proinflammatory cytokines (IL-1, IL-6, IL-8 and TNF-α), chemokines, antiapoptotic factors and stress factors [77]. A better understanding of inflammatory responses by TLR/IL-1R signaling will enable design of effective therapies for numerous debilitating chronic inflammatory diseases. Some novel strategies for anti-inflammatory therapy are achieved by interfering with the function of TIR domain of members of the TLR/IL-1R superfamily.
Inhibition of TLR/IL-1R signaling pathway components is one approach to modulation of TLR and IL-1R activity [78]. However, the high degree of cross-talk between TLR-initiated signaling pathways can enable the host immune system to overcome blocking [79]. For instance, mutation in the DD or TIR domain of MyD88, which is the main signal transducer in the TLR/IL-1R signaling pathway, does not suppress its function. Patients with MyD88 mutations are normally resistant to common microbial infections, but vulnerable to Streptococcus pneumoniae, Staphylococcus aureus, and Pseudomonas aeruginosa [80]. Similar findings were observed for IRAK-4-deficient patients [81]. Although these deficiencies are life-threatening in childhood, they become less severe with age. This also indicates that innate immunity is very important upon first encounter with pathogens. Thereafter, adaptive immunity will function even if the TLR signaling components are absent [82].
TLR/IL-1R signaling is required for innate immunity against enormous microbial infections and tissue repair through inflammatory responses. These pathways have tightly regulated activation, which otherwise might cause various inflammatory and autoimmune diseases. Some therapeutic drugs target the TIR domain of these signaling components and control hyperactivation of TLR/IL-1R signaling pathways, thereby preventing or treating human inflammatory or autoimmune diseases [83–85]. These therapeutic agents act as inhibitors and interfere with protein–protein interaction of adaptor–adaptor complexes or adaptor-TLR complexes. Targeting TIR domain is essential to blocking its downstream signaling cascade [51, 86, 87]. Some decoy peptides and synthetic inhibitory molecules are targeted to the TIR domain to interfere with protein–protein interactions in TLR/IL-1R signaling. Horng et al. [37] determined the function of TIRAP by inhibiting TIRAP using the dominant-negative mutant, TIRAP-P125H, which could not interact with TLR4 and activate TLR4 signaling. A synthetic TIRAP peptide that corresponds to the region of murine TIRAP connected to cell-permeating Antennapedia homeodomain blocks TLR4 signaling. Dimerization of the TLR4 TIR domains initiates intracellular signaling. The rational design of therapeutics to target the TLR4 TIR dimerization interface blocks TLR4 signaling. Each decoy peptide (4R1, 4R3, 4BB, 4R9, and 4αE) was synthesized in tandem with a cell-permeating Antennapedia homeodomain sequence and targeted for its inhibition of early cytokine mRNA expression and MAPK activation in LPS-stimulated primary murine macrophages. The area between BB loop of TLR4 and its fifth helical region mediate TLR4 TIR dimerization was confirmed by decoy peptides 4R1 (linker to the transmembrane region), 4BB (βB, BB and αB region), and 4αE (αE region). The emerging field of peptidomimetics is in which lead molecules mimics a functional protein epitope [88]. Targeting the BB-loop of TIR domain is also important to inhibition of MyD88-mediated signaling in vivo. The TIR domain is important in homodimerization of MyD88, and mimicking the BB-loop of the MyD88-TIR domain using synthetic epta-peptide (ST 2348) (RDVLPGT) was found to inhibit MyD88 homodimerization and IL-1 signaling in an in vitro cell system [89]. Hemorrhagic shock/resuscitation (HS/R) promotes the development of multiorgan dysfunction due to exaggerated inflammatory response. MyD88 homodimerization inhibitory peptide suppressed HMGB1/TLR4-induced IL-23 release by inhibiting IRAK4 activation in alveolar macrophages [90]. This inhibitory peptide diminished expression of the metalloproteinase-13 (MMP-13) gene, promoter activity, phosphorylation of MAPKs, and c-Jun and AP-1 activity, suggesting that MyD88 protein is a therapeutic target for arthritis-associated cartilage loss by MMP-13 [91]. Biodegradable poly(γ-glutamic acid) (γ-PGA) nanoparticles induce dendritic cell maturation. Treatment of DC with γ-PGA containing p38 MAPK inhibitor SC68376 suppressed both LPS- and NP-induced TNF-α production. MyD88 inhibitor peptide also significantly reduced TNF-α production in both LPS- and NP-treated DCs [92]. Toshchakov et al. designed cell-penetrating peptides comprising the antennapedia homeodomain of Drosophila fused with BB loop sequences of TLR2, TLR4, and TLR1/6. These TLR2- and TLR4-BB peptides (BBPs) inhibited NF-κB translocation and early IL-1β expression induced by LPS and lipopeptides [93]. They also designed a set of blocking peptides (BPs) composed of 14 amino acids corresponding to the BB loops of adapter proteins (TRAM, TRIF, MyD88, TIRAP/MAL) with a translocating sequence of the antennapedia homeodomain. These four BPs all blocked TLR4-mediated gene expression, MAPK, and transacting factor activation, but did not block TLR2-mediated activation of MAPKs. Overall, these BPs interfere with the assembly and/or stabilization of TLR4. The results also showed that, apart from the BB loop, the surfaces on MyD88 and TIRAP/MAL also enable their interaction with TLR2 [94].
TLRs and IL-1R1 are key signaling components of the innate immunity activated by microbial infections and inflammation. Bartfai et al. [95] modeled a compound based on a tripeptide sequence of the BB-loop [(F/Y)-(V/L/I)-(P/G)] of the MyD88-TIR domain and synthesized a low molecular weight MyD88 mimic compound, hydrocinnamoyl-L-valyl pyrrolidine. This cell-penetrating TIR domain mimic compound inhibited IL-1β-induced phosphorylation of p38 MAPK in EL4 thymoma cells. Pathogenesis of myocardial ischemia/reperfusion (I/R) injury is another important inflammatory disorder. Activation of NF-κB through the MyD88-dependent pathway is important for induction of innate immunity and inflammation. Inhibition of the interaction between IL-1R and MyD88 by TIR/BB-loop mimetic hydrocinnamoyl-L-valyl pyrrolidine (AS-1) attenuated myocardial ischemic injury by reducing the levels of inflammatory cytokines, adhesion molecules, myeloperoxidase activity and neutrophil infiltration in the myocardium [96]. To improve the affinity and specificity towards TIR domain, Bartfai et al. modified the inhibitor AS-1 and synthesized a novel bifunctional BB-loop TIR mimetic compounds EM77 and EM110, which disrupt the interaction of MyD88 with the IL-1R1/IL-1RAcP complex. These exhibited anti-inflammatory and neuroprotective properties through the MyD88-dependent pathway without affecting activation of protein kinase Akt/PKB, which depended on recruitment of the p85 subunit of PI3 K to IL-1R1 [97]. A peptidomimetic library of compounds was designed and tested for its inhibition of protein–protein interaction by yeast two-hybrid assay and further validated in a mammalian cell system to evaluate its inhibition of MyD88-dependent NF-κB activation [98]. One such effective compound, ST2825, is a synthetic peptide-mimetic compound modeled after the structure of a heptapeptide in the BB-loop of the MyD88-TIR domain. This compound inhibited MyD88 homodimerization of the TIR domain without influencing homodimerization of the DD domain. This ST2825 also inhibited the recruitment of IRAK1 and IRAK4 by MyD88, causing inhibition of IL-1β-mediated activation of NF-κB transcriptional activity. Thus, ST2825 is a therapeutic agent involved in the treatment of chronic inflammatory diseases by interfering with MyD88 homodimerization in TLR/IL-1R signaling [99]. RDP58 is a novel anti-inflammatory d-amino acid decapeptide that disrupts cell signaling at the pre-MAPK MyD88-IRAK-TRAF6 protein complex, inhibiting the synthesis of proinflammatory cytokines. This compound has been shown to be effective for treatment of mild-to-moderate ulcerative colitis [100].
TIR domain in apoptosis
Apoptosis is the programmed cellular death of mutated cells that exhibit irreparable DNA damage and lost control of proliferation. It is essential for maintaining tissue homeostasis, and the dysregulation of apoptosis leads to cancer [101]. It is under the influence of growth factors, hormones and cytokines, which may activate the cell death program to eliminate cells depending on the receptors present on the target cells. Apoptosis is not harmful to the host and does not induce any inflammatory responses. However, if apoptotic cells are not rapidly cleared through phagocytosis they release danger signals that provoke inflammatory responses when they proceed into secondary necrosis. In 1863, Rudolf Virchow was the first to find the link between inflammation and cancer. He suggested that lymphoreticular infiltration reflected the origin of cancer by suppressing apoptosis at the site of chronic inflammation. The inflammatory cytokines and chemokines produced by tumor cells and tumor-associated leucocytes and platelets also contribute directly to the progression of malignancy [102]. Tumor necrosis factor-alpha (TNF-α) is a major mediator of inflammation, which is involved in the destruction of diseased cells (apoptosis) at the site of inflammation and stimulates fibroblast growth. When produced chronically, TNF-α acts as an endogenous tumor promoter by tissue remodeling and stromal development, which are necessary for tumor growth and metastasis. Apoptosis of neutrophils is an important event in controlling inflammation that is accelerated by TNF-α.
Apoptosis is a host defense against pathogen invasion triggered by dsRNA, lipoproteins and lipopolysaccharides to activate TLR signaling which subsequently activates members of caspase family of cysteine proteases. Previously, Thorburn [103] reported that protein–protein interactions resulting from death domain (DD) activates effector caspases. MyD88 is the only TLR adaptor protein which can bind Fas-associated death domain protein (FADD) with its DD to activate caspase-8 for apoptosis. TIR domain containing adapter proteins also transduce signals to activate apoptosis. Out of five TIR-containing adaptor proteins, overexpression of TRIF adapter protein which has one TIR domain alone induces apoptosis through the activation of the FADD/caspase-8 axis without involving in the intrinsic pathway [104]. The TLR/IL-1R super family contains TIR domains of adapter proteins that assemble signaling components to trigger activation of transcription factors such as NF-κB and AP-1, as well as the overexpression of genes involved in immune response. However, overactivation of TIR domain mediated signaling is involved in inflammatory diseases and cancer growth [105]. The innate immune system uses the TLR/IL-1R family to signal the presence of microbial pathogens and other endogenous danger signals to the host. Aliprantis et al. [106] showed that TLR2 ligand bacterial lipoproteins (BLPs) mediate both apoptosis and the activation of NF-κB through the adaptor protein, MyD88. Inhibition of NF-kB downstream signaling pathways activates the apoptotic signaling pathway by binding of the FADD to MyD88, which subsequently activates caspase 8 (Fig. 3). Concomitantly, caspase 1 was activated by BLP-induced apoptosis, which cleaves pro-IL-1β to its mature IL-1β, a potent pro-inflammatory cytokine that is released from dying cells to generate inflammatory signals during infection [107]. Interleukin-1β, which is one of the most important inflammatory mediators, causes pancreatic islet dysfunction and apoptosis through upregulation of the expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) [108].
In addition to TLR, IL-1R carries the TIR domain in its cytoplasmic tail. IL-1 cytokine and nitric oxide (NO) have been implicated in the pathogenesis of insulin-dependent diabetes mellitus (IDDM). In vitro studies showed that islets infiltrating macrophages produce IL-1, which exhibits cytotoxicity toward β-cells by increasing the formation of NO, ceramide, prostaglandins, heat-shock proteins and protease while decreasing insulin gene expression and cyclic AMP synthesis [109]. Peptides or inhibitors that block β-cell IL-1 receptors and NO synthesis improve β-cell function in IDDM. IL-1β is also involved in apoptotic neurodegeneration in the brain. IL-1 receptor antagonist (IL-1ra) blocks the action of IL-1 and reduces ischemic and excitotoxic brain damage [110]. Another important target for AP-1 effects on cell life and death is tumor suppressor protein, for which expression and transcriptional activity are modulated by AP-1 proteins [111]. Non-canonical IKKs are involved in cancer cell survival; therefore, they are also targets for cancer therapy [71].
Conclusions
In this review, we summarized the role of the TIR domain of TLR/IL-1 receptor superfamily, which has emerged as an important participant in inflammation and apoptosis. The signaling pathways triggered by the activation of TLR/IL-1 receptor and the subsequent protein–protein interactions involve the activation of NF-κB and stress activated c-Jun N-terminal kinases (JNKs) and p38 MAP kinases, resulting in inflammatory responses. The surrounding cells adjacent to inflamed regions undergo apoptotic cell death, but this can provoke inflammatory responses when the dying cells proceed into secondary necrosis. Several reports of the three-dimensional structure of the TIR domain of human TLR/IL-1 receptors and cytosolic adapter proteins have guided researchers on the development of specific antagonists that function by inhibiting assembly of the signaling complex, and this domain has shown to have therapeutic feasibility for the treatment of inflammatory and autoimmune diseases and cancer.
References
Bonardi V, Cherkis K, Nishimura MT, Dangl JL (2012) A new eye on NLR proteins: focused on clarity or diffused by complexity? Curr Opin Immunol 24(1):41–50
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11(5):373–384
Eisenacher K, Krug A (2012) Regulation of RLR-mediated innate immune signaling – it is all about keeping the balance. Eur J Cell Biol 91(1):36–47
Kingeter LM, Lin X (2012) C-type lectin receptor-induced NF-κB activation in innate immune and inflammatory responses. Cell Mol Immunol 9(2):105–112
Werts C, Girardin SE, Philpott DJ (2006) TIR, CARD and PYRIN: three domains for an antimicrobial triad. Cell Death Diff 13(5):798–815
Janeway CA Jr, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20:197–216
Akira S, Takeda K, Kaisho T (2001) Toll-like receptors: critical proteins linking innate and acquired immunity. Nature Immunol 2(8):675–680
Akira S, Takeda K (2004) Toll-like receptors signalling. Nat Rev Immunol 4(7):499–511
Rakoff-Nahoum S, Medzhitov R (2009) Toll-like receptors and cancer. Nat Rev Cancer 9(1):57–63
Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA (1996) The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86(6):973–983
Williams MJ, Rodriguez A, Kimbrell DA, Eldon ED (1997) The 18-wheeler mutation reveals complex anti-bacterial gene regulation in Drosophila host defense. EMBO J 16(20):6120–6130
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr (1997) A human homo-logue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388:394–397
Kawai T, Akira S (2009) The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol 21(4):317–337
Beutler BA (2009) TLRs and innate immunity. Blood 113(7):1399–1407
Jin MS, Lee JO (2008) Structures of the Toll-like receptor family and its ligand complexes. Immunity 29(2):182–191
Iwasaki A, Medzhitov R (2004) Toll-like receptor control of the adaptive immune responses. Nat Immunol 5(10):987–995
Dai J, Liu B, Li Z (2009) Regulatory T cells and Toll-like receptors: what is the missing link? Int Immunopharmacol 9(5):528–533
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124(4):783–801
Miyake K (2007) Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin Immunol 19(1):3–10
Kumar H, Kawai T, Akira S (2009) Toll-like receptors and innate immunity. Biochem Biophys Res Commun 388(4):621–625
Kim YM, Brinkmann MM, Paquet ME, Ploegh HL (2008) UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 452(7184):234–238
Bowie A, O’Neill LAJ (2000) The interleukin-1 receptor/Toll-like receptor superfamily: signal generators for pro-inflammatory interleukins and microbial products. J Leukoc Biol 67(4):508–514
Greenfeder SA, Nunes P, Kwee L, Labow M, Chizzonite RA, Ju G (1995) Molecular cloning and characterization of a second subunit of the interleukin 1 receptor complex. J Biol Chem 270(23):13757–13765
Gay N, Keith F (1991) Drosophila Toll and IL-1 receptor. Nature 351(6325):355–356
Dinarello CA (2009) Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27:519–550
Subramaniam S, Stansberg C, Cunningham C (2004) The interleukin 1 receptor family. Dev Comp Immunol 28(5):415–428
Heguy A, Baldari CT, Macchia G, Telford JL, Melli M (1992) Amino acids conserved in interleukin-1 receptors (IL-1Rs) and the drosophila Toll protein are essential for IL-1R signal transduction. J Biol Chem 267(4):2605–2609
Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, Gorman DM, Bazan JF, Kastelein RA (2005) IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23(5):479–490
Boraschi D, Tagliabue A (2006) The interleukin-1 receptor family. Vitam Horm 74:229–254
Burns K, Janssens S, Brissoni B, Olivos N, Beyaert R, Tschopp J (2003) Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J Exp Med 197(2):263–268
Lord KA, Hoffman-Liebermann B, Liebermann DA (1990) Nucleotide sequence and expression of a cDNA encoding MyD88, a novel myeloid differentiation primary response gene induced by IL6. Oncogene 5(7):1095–1097
Kawai T, Adachi O, Ogawa T, Takeda K, Akira S (1999) Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11(1):115–122
Takeuchi O, Takeda K, Hoshino K, Adachi O, Ogawa T, Akira S (2000) Cellular responses to bacterial cell wall components are mediated through MyD88-dependent signaling cascades. Int Immunol 12(1):113–117
Hacker H, Vabulas RM, Takeuchi O, Hoshino K, Akira S, Wagner H (2000) Immune cell activation by bacterial CpG-DNA through myeloid differentiation marker 88 and tumor necrosis factor receptor-associated factor (TRAF)6. J Exp Med 192(4):595–600
Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A (2001) The innate immune response to bacterial flagellin is mediated by Toll-like receptor-5. Nature 410(6832):1099–1103
Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo S, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S (2002) Small antiviral compounds activate immune cells via TLR7 MyD88-dependent signaling pathway. Nat Immunol 3(2):196–200
Horng T, Barton GM, Medzhitov R (2001) TIRAP: an adapter molecule in the Toll signaling pathway. Nat Immunol 2(9):835–841
Yamamoto M, Sato S, Hemmi H, Sanjo H, Uematsu S, Kaisho T, Hoshino K, Takeuchi O, Kobayashi M, Fujita T, Takeda K, Akira S (2002) Essential role of TIRAP/Mal for activation of the signaling cascade shared by TLR2 and TLR4. Nature 420(6913):324–329
Horng T, Barton GM, Flavell RA, Medzhitov R (2002) The adaptor molecule TIRAP provides signaling specificity for Toll-like receptors. Nature 420(6913):329–333
Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S (2003) Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science 301(5633):640–643
Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B (2003) Identification of Lps2 as a key transducer of MyD88-independnet TIR signaling. Nature 424(6950):743–748
Bin LH, Xu LG, Shu HB (2003) TIRP, a novel Toll/interleukin-1 receptor (TIR) domain-containing adapter protein involved in TIR signaling. J Biol Chem 278:24526–245312
Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, Takeuchi O, Takeda K, Akira S (2003) TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol 4(11):1144–1150
Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, Monks B, Pitha PM, Golenbock DT (2003) LPS-TLR4 signaling to IRF-3/7 and NF-κB involves the Toll adapters TRAM and TRIF. J Exp Med 198(7):1043–1055
Oshiumi H, Sasai M, Shida K, Fujita T, Matsumoto M, Seya T (2003) TIR-containing adapter molecule (TICAM)-2: a bridging adapter recruiting to Toll-like receptor 4 TICAM-1 that induces interferon-β. J Biol Chem 278(50):49751–49762
Mink M, Fogelgren B, Olszewski K, Maroy P, Csiszar K (2001) A novel human gene (SARM) at chromosome 17q11 encodes a protein with a SAM motif and structural similarity to Armadillo/β-catenin that is conserved in mouse, Drosophila and C. elegans. Genomics 74:234–244
O’Neill LAJ, Fitzgerald KA, Bowie AG (2003) The Toll-IL-1 receptor adaptor family grows to five members. Trends in Immunol 24(6):286–289
Carty M, Goodbody R, Schroder M, Stack J, Moynagh PN, Bowie AG (2006) The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat Immunol 7(10):1074–1081
Szretter KJ, Samuel MA, Gilfillan S, Fuchs A, Colonna M, Diamond MS (2009) The immune adaptor molecule SARM modulates tumor necrosis factor alpha production and microglia activation in the brainstem and restricts west Nile virus pathogenesis. J Virol 83(18):9329–9338
Radons J, Gabler S, Wesche H, Korherr C, Hofmeister R, Falk W (2002) Identification of essential regions in the cytoplasmic tail of interleukin-1 receptor accessory protein critical for interleukin-1 signaling. J Biol Chem 277(19):16456–16463
Xu Y, Tao X, Shen B, Horng T, Medzhitov R, Manley JL, Tong L (2000) Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature 408(6808):111–115
Nyman T, Stenmark P, Flodin S, Johansson I, Hammarstrom M, Nordlund PR (2008) The crystal structure of the human Toll-like receptor 10 cytoplasmic domain reveals a putative signaling dimer. J Biol Chem 283(18):11861–11865
Burns K, Martinon F, Esslinger C, Pahl H, Schneider P, Bodmer JL, Marco FD, French L, Tschopp J (1998) MyD88, an adapter protein involved in interleukin-1 signaling. J Biol Chem 273(20):12203–12209
Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA Jr (1998) MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell 2(2):253–258
Khan JA, Brint EK, O’Neill LAJ, Tong L (2004) Crystal structure of the Toll/Interleukin-1 receptor domain of human IL-1RAPL. J Biol Chem 279:31664–31670
Ohnishi H, Tochio H, Kato Z, Orii KE, Li A, Kimura T, Hiroaki H, Kondo N, Shirakawa M (2009) Structural basis for the multiple interactions of the MyD88 TIR domain in TLR4 signaling. Proc Natl Acad Sci USA 106(25):10260–10265
Valkov E, Stamp A, DiMaio F, Baker D, Verstak B, Roversi P, Kellie S, Sweet MJ, Mansell A, Gay NJ, Martin JL, Kobe B (2011) Crystal structure of Toll-like receptor adaptor MAL/TIRAP reveals the molecular basis for signal transduction and disease protection. Proc Natl Acad Sci USA 108(36):14879–14884
Jang TH, Park HH (2014) Crystal structure of TIR domain of TLR6 reveals novel dimeric interface of TIR-TIR interaction for Toll-like receptor signaling pathway. J Mol Biol. doi:10.1016/j.jmb.2014.07.024
Zhou K, Kanai R, Lee P, Wang HW, Modis Y (2012) Toll-like receptor 5 forms asymmetric dimers in the absence of flagellin. J Struct Biol 177(2):402–409
Lin Z, Lu J, Zhou W, Shen Y (2012) Structural insignts into TIR domain specificity of the bridging adaptor Mal in TLR4 signaling. PLoS One 7(4):e34202
Loiarro M, Gallo G, Fanto N, Santis RD, Carminati P, Ruggiero V, Sette C (2009) Identification of critical residues of the MyD88 death domain involved in the recruitment of downstream kinases. J Biol Chem 284(41):28093–28103
Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ (2000) Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103(2):351–361
Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, Chiu YH, Deng L, Chen ZJ (2004) TAB 2 and TAB 3 activate the NF-κB pathway through binding to polyubiquitin chains. Mol Cell 15(4):535–548
Carmody RJ, Chen YH (2007) Nuclear factor-κB: activation and regulation during Toll-like receptor signaling. Cell Mol Immunol 4(1):31–41
Yao J, Tae WK, Qin J, Jiang Z, Qian Y, Xiao H, Lu Y, Qian W, Gulen MF, Sizemore N, DiDonato J, Sato S, Akira S, Su B, Li X (2007) Interleukin-1 (IL-1)-induced TAK1-dependent versus MEKK3-dependent NF κB activation pathways bifurcate at IL-1 receptor-associated kinase modification. J Biol Chem 282(9):6075–6089
Sanz L, Diaz-Meco MT, Nakano H, Moscat J (2000) The atypical PKC-interacting protein p62 channels NF-κB activation by the IL-1-TRAF6 pathway. EMBO J 19(7):1576–1586
Karin M, Liu ZG, Zandi E (1997) AP-1 function and regulation. Curr Opin Cell Biol 9(2):240–246
Shaulian E, Karin M (2002) AP-1 as a regulator of cell life and death. Nat Cell Biol 4(5):E131–E136
Kawai T, Akira S (2006) TLR signalling. Cell death Differ 13(5):816–825
Wagner EF, Nebreda AR (2009) Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 9(8):537–549
Kim JY, Beg AA, Haura EB (2013) Non-canonical IKKs, IKKε and TBK1, as novel therapeutic targets in the treatment of non-small cell lung cancer. Expert Opin Ther Targets 17(10):1109–1112
Doyle SE, Vaidya SA, O’Connell R, Dadgostar H, Dempsey PW, Wu T, Rao G, Sun R, Haberland M, Modlin R, Cheng G (2002) IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 17(3):251–263
Hoshino K, Kaisho T, Iwabe T, Takeuchi O, Akira S (2002) Differential involvement of IFN-β in Toll-like receptor-stimulated dendritic cell activation. Int Immunol 14(10):1225–1231
Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN (2002) TLR4, but not TLR2, mediates IFN-β-induced STAT1α/β-dependent gene expression in macrophages. Nat Immunol 3(4):392–398
Kumar H, Takeuchi O, Akira S (2013) Toll-like receptors. In: Lennarz WJ, Lane MD (eds) Encyclopedia of biological chemistry, vol 2. Academic Press, New York, pp 396–401
Salaun B, Romero P, Lebecque S (2007) Toll-like receptors’ two-edged sword: when immunity meets apoptosis. Eur J Immunol 37(12):3311–3318
Li Q, Verma IM (2002) NF-κB regulation in the immune system. Nat Rev Immunol 2(10):725–734
Schmidt C (2006) Immune system’s Toll-like receptors have good opportunity for cancer treatment. J Natl Cancer Inst 98(9):574–575
O’Neill LAJ (2006) Targeting signal transduction as a strategy to treat inflammatory diseases. Nat Rev Drug Disc 5(7):549–563
Li F, Thiele I, Jamshidi N, Palsson BO (2009) Identification of potential pathway mediation targets in Toll-like receptor signaling. PLoS Comp Biol 5(2):e1000292
Von Bernuth H, Picard C, Jin Z et al (2008) Pyogenic bacterial infections in humans with MyD88 deficiency. Science 321(5889):691–696
Ku CL, von Bernuth H, Picard C et al (2007) Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J Exp Med 204(10):2407–2422
Marsh BJ, Stenzel-Poore MP (2008) Toll-like receptors: novel pharmacological targets for the treatment of neurological diseases. Curr Opin Pharmacol 8(1):8–13
O’Neill LAJ, Bryant CE, Doyle SL (2009) Therapeutic targeting of Toll-like receptors for infectious and inflammatory diseases and cancer. Pharmacol Rev 61(2):177–197
Pedras-Vasconcelos J, Puig M, Verthelyi D (2009) TLRs as therapeutic targets in CNS inflammation and infection. Front Biosci (Elite Ed) 1:476–487
Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282(5396):2085–2088
Li C, Zienkiewicz J, Hawiger J (2005) Interactive sites in the Myd88 Toll/interleukin (IL) 1 receptor domain responsible for coupling to the IL1β signaling pathway. J Biol Chem 280:26152–26159
Toshchakov VV, Szmacinski H, Couture LA, Lakowicz JR, Vogel SN (2011) Targeting TLR4 signaling by TLR4 TIR-derived decoy peptides: identification of the TLR4 TIR dimerization interface. J Immunol 186(8):4819–4827
Loiarro M, Sette C, Gallo G, Ciacci A, Fanto N, Mastroianni D, Carminati P, Ruggiero V (2005) Peptide-mediated interference of TIR domain dimerization in MyD88 inhibits interleukin-1-dependent activation of NF-κB. J Biol Chem 280(16):15809–15814
Liu Y, Yuan Y, Li Y, Zhang J, Xiao G, Vodovotz Y, Billiar TR, Wilson MA, Fan J (2009) Interacting neuro-endocrine, innate, and acquired immune pathways regulate neutrophil mobilization from bone marrow following hemorrhagic shock. J Immunol 182(1):572–580
Ahmad R, Sylvester J, Zafarullah M (2007) MyD88, IRAK1 and TRAF6 knockdown in human chondrocytes inhibits interleukin-1-induced matrix metalloproteinase-13 gene expression and promoter activity by impairing MAP kinase activation. Cell Signal 19(12):2549–2557
Uto T, Wang X, Sato K, Haraguchi M, Akagi T, Akashi M, Baba M (2007) Targeting of antigen to dendritic cells with poly(γ-glutamic acid) nanoparticles induces antigen-specific humoral and cellular immunity. J Immunol 178(5):2979–2986
Toshchakov VY, Fenton MJ, Vogel SN (2007) Cutting edge: differential inhibition of TLR signaling pathways by cell-permeable peptides representing BB loops of TLRs. J Immunol 178(5):2655–2660
Toshchakov VU, Basu S, Fenton MJ, Vogel SN (2005) Differential involvement of BB loops of Toll-IL-1 resistance (TIR) domain-containing adapter proteins in TLR4- versus TLR2-mediated signal transduction. J Immunol 175(1):494–500
Bartfai T, Behrens MM, Gaidarova S, Pemberton J, Shivanyuk A, Rebek J Jr (2003) A low molecular weight mimic of the Toll/IL-1 receptor/resistance domain inhibits IL-1 receptor-mediated responses. Proc Natl Acad Sci USA 100(13):7971–7976
Cao Z, Hu Y, Wu W, Ha T, Kelley J, Deng C, Chen Q, Li C, Li J, Li Y (2009) The TIR/BB-loop mimetic AS-1 protects the myocardium from ischaemia/reperfusion injury. Cardiovasc Res 84(3):442–451
Davis CN, Mann E, Behrens MM, Gaidarova S, Rebek M, Rebek J Jr, Bartfai T (2006) MyD88-dependent and –independent signaling by Il-1 in neurons probed by bifunctional Toll/IL-1 receptor domain/BB-loop mimetics. Proc Natl Acad Sci USA 103(8):2953–2958
Fanto N, Gallo G, Ciacci A, Semproni M, Vignola D, Quaglia M, Bombardi V, Mastroianni D, Zibella MP, Basile G, Sassano M, Ruggiero V, De Santis R, Carminati P (2008) Design, synthesis, and in vitro activity of peptidomimetic inhibitors of myeloid differentiation factor 88. J Med Chem 51(5):1189–1202
Loiarro M, Capolunghi F, Fanto N, Gallo G, Campo S, Arseni B, Carsetti R, Carminati P, De Santis R, Ruggiero V, Sette C (2007) Pivotal advance: inhibition of Myd88 dimerization and recruitment of IRAK1 and IRAK4 by a novel peptidomimetic compound. J Leukoc Biol 82(4):801–810
Travis S, Yap LM, Hawkey C, Warren B, Lazarov M, Fong T, Tesi RJ (2005) RDP58 is a novel and potentially effective oral therapy for ulcerative colitis. Inflamm Bowel Dis 11(8):713–719
Kerr JFR, Wyllie AH, Currie AR (1972) Apoptosis: basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26(4):239–257
Balkwill F, Mantovani A (2001) Inflammation and cancer: back to Virchow? Lancet 357:539–545
Thorburn A (2004) Death receptor-induced cell killing. Cell Signal 16(2):139–144
Kaiser WJ, Offermann MK (2005) Apoptosis induced by the Toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J Immunol 174(8):4942–4952
Fekonja O, Avbelj M, Jerala R (2012) Suppression of TLR signaling by targeting TIR domain-containing proteins. Curr Protein Pept Sci 13(8):776–788
Aliprantis AO, Yang RB, Weiss DS, Godowski P, Zychlinsky A (2000) The apoptotic signaling pathway activated by Toll-like receptor-2. EMBO J 19(13):3325–3336
Colomar A, Marty V, Medina C, Combe C, Parnet P, Amedee T (2003) Maturation and release of interleukin-1β by lipopolysaccharide-primed mouse Schwann cells require the stimulation of P2X7 receptors. J Biol Chem 278(33):30732–30740
Barshes NR, Wyllie S, Goss JA (2005) Inflammation-mediated dysfunction and apoptosis in pancreatic islet transplantation: implications for intrahepatic grafts. J Leukoc Biol 77(5):587–597
Sjoholm A (1998) Aspects of the involvement of interleukin-1 and nitric oxide in the pathogenesis of insulin-dependent diabetes mellitus. Cell Death Differ 5(6):461–468
Rothwell N, Allan S, Toulmond S (1997) The role of interleukin 1 in acute neurodegeneration and stroke: pathophysiolocial and therapeutic implications. J Clin Invest 100(11):2648–2652
Shaulian E, Karin M (2001) AP-1 in cell proliferation and survival. Oncogene 20(19):2390–2400
Acknowledgments
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) of the Ministry of Education, Science and Technology (2012R1A2A2A01010870).
Conflict of interest
The authors have no conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Narayanan, K.B., Park, H.H. Toll/interleukin-1 receptor (TIR) domain-mediated cellular signaling pathways. Apoptosis 20, 196–209 (2015). https://doi.org/10.1007/s10495-014-1073-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-014-1073-1