
Abstract
PDCD5 (programmed cell death 5) accelerates apoptosis of certain tumor cells and the replication-defective Ad-PDCD5 may be a promising agent for enhancing chemosensitivity. In this study, a triple-regulated conditionally replicating adenoviruses (CRAd) carrying PDCD5 gene expression cassette, SG611-PDCD5, was engineered. In SG611-PDCD5, the E1a gene with a deletion of 24 nucleotides within CR2 region is controlled under the human telomerase reverse transcriptase (hTERT) promoter, the E1b gene expression is directed by the hypoxia response element (HRE), whereas the PDCD5 gene is controlled by the cytomegalovirus promoter. The tumor-selective replication of this virus and its antitumor efficacy were characterized in several leukemic cell lines in vitro and in xenograft models of human leukemic cell line in nude mice. It was found by RQ-RT-PCR assay that SG611-PDCD5 expressed PDCD5 efficiently in leukemic cells. In K562 tumor xenograft models, SG611-PDCD5 displayed a tumor killing capacity. At a dose of 1 × 109 plaque-forming units, SG611-PDCD5 alone could completely inhibit the tumor growth and more effective than replication-defective Ad-PDCD5. Histopathologic examination revealed that SG611-PDCD5 administration resulted in leukemic cell apoptosis. We concluded that the triple-regulated SG611-PDCD5, as a more potent and safer antitumor therapeutic, could provide a new strategy for leukemia biotherapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Recent advances in understanding the molecular pathogenesis of cancer and the rapid development of recombinant DNA technology have made cancer gene therapy feasible in the clinical setting. Apoptosis-related genes such as p53, p73, Bax and PDCD5 [1–4] have been used in gene therapy to directly induce apoptosis or by synergistically enhancing the effects of chemotherapy in cancer cells.
However, traditional gene therapy showed the low transfection, no specificity, and poor efficacy. Conditionally replicating adenoviruses (CRAd) can replicate specifically in cancer cells and lyse them. When the CRAd vectors are armed with the antitumor transgene, the transgene copies and expression may be increased markedly along with the preferable replication of CRAds in tumor cells but not in normal cells, so that the antitumor efficacy was enhanced [5]. Thereby, the CRAds overcome the disadvantages of traditional gene therapy and are emerging as powerful tools in the warfare on cancer. The ability to modify cell-specific infectivity or tissue-specific replication machinery, as well as the possibility of modifying viral-cellular protein interactions with cellular checkpoint regulators are emerging as new trends in the design of safer and more effective adenoviruses [6].
Based on two common characteristics of telomerase activation and low oxygen tension environment in most cancers, a human telomerase reverse transcriptase (hTERT) promoter-regulated CRAd [7, 8], CNHK300 with the E1a gene controlled by the hTERT promoter, and a dual-regulated CRAd, CNHK500, with the E1a and E1b genes controlled by the hTERT promoter and the hypoxia response element (HRE) [9], respectively, were reported previously. They showed that the simultaneous regulation of E1a and E1b transcription by two promoters could greatly increase the tumor specificity of CRAds and reduce adverse side effects in normal cells.
Recently, a triple-regulated CRAd carrying p53 gene expression cassette, SG600-p53, was reported [10]. In SG600-p53, the E1a gene with a deletion of 24 nucleotides within CR2 region is controlled under the hTERT promoter, the E1b gene expression is directed by the HRE, whereas the p53 gene is controlled by the cytomegalovirus promoter. SG600-p53 displayed a tumor-selective killing capacity and more effective than replication-defective Ad-p53.
PDCD5 (programmed cell death 5), also designated TF-1 cell apoptosis-related gene-19 (TFAR19), was identified in TF-1 cells undergoing apoptosis [11]. Functional studies indicate that recombinant human PDCD5 accelerates apoptosis of tumor cells. PDCD5 is involved in paraptosis, another form of programmed cell death that may occur during carcinogenesis, and neurodegeneration [12–14]. PDCD5 is expressed in lower levels in the hepatocellular carcinoma, breast cancer, gastric cancer, astrocytic gliomas and leukemia [15–21], suggesting that reduced PDCD5 expression may play an important role in the mechanism of cancer pathogenesis. Ad-PDCD5 has been used in gene therapy by synergistically enhancing the effects of chemotherapy in cancer cells [4].
In the study, SG611-PDCD5, a triple-regulated CRAd carrying PDCD5 gene expression cassette, was constructed based on the SG600 system. We believe that not only the expression of PDCD5 gene is increased markedly but also the antitumor effect of the generated SG611-PDCD5 is improved.
Materials and methods
Cell lines and culture conditions
Human chronic myelogenous leukemia cell lines K562, human acute promyelocytic leukemia cell line NB4 and human megakaryoblastic leukemia cell line Dami and MEG-01, were kindly provided by Cell Bank Shanghai Institute of Cell Biology (Shanghai, China). Human embryo kidney cell line (HEK293), human fibroblast cell line BJ, and human hepatic cell L-02 were purchased from the American Type Culture Collection. All cell lines were cultured according to the instructions of the providers.
Virus preparation
The construction, generation, and purification of Ad-PDCD5 as well as the identification of TCID50 (50% tissue culture infective dose, TCID50) were performed by the Applied Gene Technology Corporation Ltd. (Beijing, China). The Ad-PDCD5 vector has the E1 and E3 subunits deleted, and contains the PDCD5 transgene under the control of the CMV promoter.
The vector pSG600, with the E1a and E1b genes controlled by the hTERT promoter and the HRE, respectively, was reported previously [10] and was kindly provided by the Laboratory of Viral and Gene Therapy, Eastern Hepatobiliary Surgical Hospital (Shanghai, China). The plasmids pClon15, PENTR11-Linker, and pPE3-F11B were purchased from the Microbix Biosystems Inc. (Toronto, Ontario, Canada).
The plasmid pDC316-PDCD5 was kindly provided by the Prof. Da-Long Ma (Peking University, Beijing). Firstly, PDCD5 gene was released from pDC316-PDCD5 using EcoRI digestion. After sequencing confirmation, the PDCD5 cDNA was cloned into plasmid pClon15 by EcoRI in forward orientation, which has AgeI and NotI site. Secondly, the PDCD5 fragment with mCMVp and SV40 polyA was released from pClon15 and ligated with an PENTR11-Linker by using Age I and NotI. The obtained novel adenoviral vector, named PENTR11-PDCD5, was then ligated with pPE3-F11B, which is the backbone of the adenovirus with fiber knob of adenovirus serotype 11B. A novel plasmid pPE3-F11B-PDCD5 was then generated. Lastly, the plasmid pPE3-F11B-PDCD5 was co-transfected with pSG600 into HEK293 cells using Lipofectamine 2000 (Qiagen) to construct the combinant adenovirus. After homologously recombining in HEK293 cells, the CRAd, named SG611-PDCD5, was obtained. Similarly, the CRAd vector SG611 and the CRAd vector SG611-EGFP containing the enhanced green fluorescent protein were constructed as a null control, and a marker control, respectively.
Replication, purification and titer determination
HEK293 cells were infected with SG611-PDCD5, SG611 and SG611-EGFP incubated at 37°C under the condition of 5% CO2. Upon replicating to adequate volume in 293 cells, the virus was purified by a routine method with CsCl2. Permissive HEK293 cells were seeded in 96-well plates (104 cells/well). The next day, eight serial tenfold dilutions of purified viruses were subjected to 96-well plates. Seven days after incubation, the observable cytopathic effect (CPE) per dilution was counted and virus titer was determined with the TCID50 method [22].
Flow cytometry analysis
To investigate the infection efficiency of SG611 vector, K562 cells were harvested in the exponential growth phase. The cells were seeded in six-well plates at a density of 8 × 105 per well and infected with SG611-EGFP at a MOI (multiplicity of infection, MOI) of 1, 5 and 10 pfu (plaque-forming units, pfu)/cell. Mock infection with normal saline was as control. The cells were co-cultured for 6, 24, 48, 72, 96 and 120 h and then washed to remove free viral particles. The percentage of EGFP positive cells was analyzed on a FACS Sort flow cytometer (Becton, Dickinson and Company, Mountainview, CA, US) with excitation at 488 nm using an excitation wave argon ion laser.
RQ-PCR for measurement of relative PDCD5 expression levels
In the in vitro experiments, K562 cells were seeded in six-well plates at a density of 8 × 105/per well. To investigate the relation of time and PDCD5 expression, the cells were infected with SG611-PDCD5, SG611-EGFP, Ad-PDCD5 and control with normal saline at a MOI of 1 pfu/cell, respectively. At days 0, 1, 2, 3, 4, 5, 6, 7, 8 and day 9 after infection, the cells were collected and total cellular RNA was extracted using the TRIzol® reagent (Invitrogen, US) as described by the manufacturer. The cDNA synthesis reaction was performed and was used to detect PDCD5 expression by RQ-PCR as described previously [20].
In vitro cell viability
Cell viability was measured by MTT (methyl thiazolyl tetrazolium, MTT) assay to evaluate the cytotoxicity of adenovirus vectors. A panel of leukemic cells was planted at a density of 1 × 104 per well in 96-well plates. After incubation for 24 h, the cells were infected with SG611-PDCD5, SG611, Ad-PDCD5 or mock infected as controls at a gradient of MOI from 0 to 320 pfu/cell. The MTT assay was separately performed after 48, 96 and 168 h incubation. 20 μl of a 5 mg/ml solution of MTT (Sigma, US) was added to wells and incubated for additional 4–6 h at 37°C. The reaction was stopped with 0.01 M HCL in 10% SDS, 100 μl per well. The absorbance was determined on a plate reader read at 570 nm on a scanning multiwell pectrophotometer with a reference wavelength at 655 nm (Bio-Rad650, US). Each assay was repeated three times. Cell viability curves were generated by Excel computer software.
Animal experiments
BALB/c nu/nu female mice (4–5 weeks of age; Experimental Animal Center, Peking University Health Science center, Beijing, China) were used in this study. All care and handling of animals were performed with the approval of Institutional Authority for Laboratory Animal Care of Peking University. Mice were pretreated by intraperitoneal (i.p.) injections of cyclophosphamine once daily at a dose of 100 mg/kg for two consecutive days. Two days later, a total of 1.5 × 107 K562 cells in a total volume of 100 μl were s.c. injected into the right flank of the mice. When the diameter of the tumor exceeded 5–7 mm, mice were randomly divided into four groups, as follows: 5 mice for Group I, Ad-PDCD5; 6 mice for Group II, SG611; 6 mice for Group III, SG611-PDCD5 and 5 mice for control group. Mice were given total 1 × 109 pfu viruses by five times of intratumoral injections, one time every other day, in the Ad-PDCD5 Group I, SG611 Group II and SG611-PDCD5 group III, respectively. In the control group, 100 μl normal saline per mouse per time was injected intratumorally. Tumor sizes were monitored every other day. Tumor volume was calculated using the following formula: (maximal diameter) × (perpendicular diameter)2 × 0.52.
TUNEL assay
Mice were sacrificed 28 days later according to the institutional guidelines. The tumors were removed, fixed in 10% neutral formaldehyde for 6 h, and embedded with paraffin. Tumor sections were subjected to immunohistochemistry. TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay apoptosis was detected in tumor tissues using the TUNEL kit (Intergen Co. Ltd., Burlington, MA, US) according to instructions provided by the manufacturer. Briefly, samples were digested with 200 mg/l proteinase K at 37°C for 20 min. A volume of 20 μl kit reaction agent was added to each sample according to the kit manual and paraffin-covered slides were incubated at 37°C for 60 min in a humidified chamber. An aliquot of 2 μl terminal deoxynucleotidyl transferase and 18 μl nucleotide mixture containing biotind UTP were added to the reactions. The slides were washed twice with phosphate buffer, and each slide was incubated with 50 μl SP-AP (Promega, Madison, WI, US) at 37°C for 1 h. Sections were visualized with BCIP/NBT (5-bromo-4-chloro-3-indolyl phosphate/nitro bluetetrazolium) using a light microscope, and nuclei of positive cells appeared blue–black. At 250× magnification, the eyepiece provided a field size of 0.72 mm2. Results are expressed as the mean apoptotic cells percentage, and three different fields per slide were examined.
Statistical analysis
Data from in vitro experiments were assessed by Student’s t test and data from in vivo experiments were assessed by ANOVA. Findings of P < 0.05 were considered significant.
Results
SG611-PDCD5 adenovirus
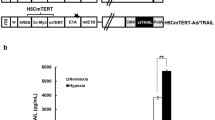
As showed in Fig. 1, In SG611, the hTERT promoter and HRE were used to regulate the E1a and E1b genes, respectively. A PDCD5 gene expression cassette containing cytomegalovirus promoter, PDCD5 cDNA, and SV40 polyA was inserted into the genome of SG611 and then generated SG611-PDCD5. The generation and purification of SG611-PDCD5 as well as the identification of TCID50 was performed. The titers of the viral stocks were determined to be 2.91 × 1010, 2.06 × 1010, 7.89 × 109 and 2.20 × 1010 TCID50/ml for SG611-PDCD5, SG611, SG611-EGFP and Ad-PDCD5, respectively.

Schematic diagram of the SG611-PDCD5 adenoviral construct. In SG611, the hTERT promoter and HRE were used to regulate the E1a and E1b genes, respectively. A PDCD5 gene expression cassette containing cytomegalovirus promoter, PDCD5 cDNA, and SV40 polyA was inserted into the genome of SG611 and then generated SG611-PDCD5. ITR, inverted terminal repeats; ψ, adenovirus type 5 packaging signal
Flow cytometry analysis for the infection efficiency of SG611
The K562 cells were seeded in six-well plates at a density of 8 × 105 per well and infected with SG611-EGFP at a MOI of 1, 5 and 10 pfu/cell. The cells were co-cultured for 24, 48, 72, 96 and 120 h and the percentage of EGFP positive cells was analyzed on a FACS Sort flow cytometer. The results showed that the infection efficiency was increased gradually along with the increase of MOI (Fig. 2). At 24 h after infection the percentage of EGFP positive cells with a MOI of 1, 5 and 10 pfu/cell was 6.39, 46.47 and 71.16%, respectively. At a MOI of 1, 5 or 10, the percentage of EGFP positive cells was also increased gradually along with time prolonging. At a MOI of 5, the percentage of EGFP positive cells for 24, 48, 72, 96 and 120 h was 46.47, 55.44, 91.27, 95.38 and 97.74%, respectively.

The percentage of EGFP positive cells for K562 was analyzed on a FACS Sort flow cytometer. The results showed that the infection efficiency was increased gradually along with the increase of MOI or time prolonging
SG611-PDCD5-mediated PDCD5 expression
The K562 cells was cultured in six-well plates at a density of 8 × 105 per well and infected with SG611-PDCD5, SG611-EGFP and Ad-PDCD5 at a MOI of 1 pfu/cell, respectively. At 0–9 days after infection, the cells were collected, total cellular RNA was extracted, cDNA synthesis reaction was performed and was used to detect PDCD5 expression by RQ-PCR as described previously [20]. The results showed that SG611-PDCD5 expressed PDCD5 with high efficiency in cancer cells compared with Ad-PDCD5. At a MOI of 1 pfu/cell, the expression level of PDCD5 was increased gradually along with time prolonging (Fig. 3). The ratio of PDCD5/ABL at days 4, 5, 6, 7, 8 and day 9 was 3.53 ± 0.65, 11.45 ± 1.75, 33.14 ± 11.43, 39.23 ± 16.85, 70.12 ± 3.13 and 168.32 ± 32.13, respectively.

Transgene expression by SG611-PDCD5. The results showed that SG611-PDCD5 expressed PDCD5 with high efficiency in K562 cells compared with Ad-PDCD5. At a MOI of 1 pfu/cell, the expression level of PDCD5 was increased gradually along with time prolonging
SG611-PDCD5-mediated cancer cell-killing effect
For assessing the killing effect of adenovirus vectors, a panel of leukemic cells was planted at a density of 1 × 104 per well in 96-well plates. The cells were infected with SG611-PDCD5, SG611, Ad-PDCD5 or mock infected as controls at a gradient of MOI from 0 to 320 pfu/cell. The MTT assay was separately performed after 48, 96 and 168 h incubation. The results showed that the cell viabilities were decreased gradually along with the increase of MOI in these leukemic cells with SG611-PDCD5 in comparison with SG611 and Ad-PDCD5 (Fig. 4). At 168 h after infection at a MOI of 20, 40 and 80 pfu/cell, the cell viability of K562 cells was lower in the group treated with SG611-PDCD5 than that with SG611 or Ad-PDCD5 (all P < 0.01). Similar results were found in the following cell lines: MEG-01 at 168 h after infection at a MOI of 20, 40 and 80 pfu/cell, respectively; Dami at 48 or 96 h after infection at a MOI of 10, 40, 80, 160 and 320 pfu/cell, respectively; NB4 at 96 h after infection at a MOI of 40, 80, 160 and 320 pfu/cell, respectively (all P < 0.01). However, the cell viabilities were maintained a relatively higher level in normal cell lines BJ (about 60%) and L-02 (about 90%) with SG611-PDCD5 even at a MOI of 1,000 and 1,280 pfu/cell, respectively.

Cytotoxic effects in a panel of leukemic cells after infection with SG611-PDCD5 in comparison with SG611 and Ad-PDCD5 at escalating MOI value by MTT assay. a K562 at 168 h; b MEG-01 at 168 h; c Dami at 48 h; d Dami at 96 h; e NB4 at 96 h; f human fibroblast cell line BJ at 96 h; g human hepatic cell line L-02 at 96 h; h L-02 at 168 h. The results showed that the cell viabilities were decreased gradually along with the increase of MOI in these leukemic cells with SG611-PDCD5 in comparison with SG611 and Ad-PDCD5. However, the cell viabilities were maintained a relatively higher level in normal cell lines BJ (about 60%) and L-02 (about 90%) with SG611-PDCD5 even at a MOI of 1,000 and 1,280 pfu/cell, respectively
Antitumor efficacy of SG611-PDCD5 on tumor xenografts
We evaluated the antitumor efficacy of SG611-PDCD5 on K562 tumor xenografts established in nude mice. Mice were given total 1 × 109 pfu viruses by five times of intratumoral injections, one time every other day, in the Ad-PDCD5 Group I, SG611 Group II and SG611-PDCD5 group III, respectively. Tumor sizes were monitored every other day. Tumor volume was calculated using the following formula: (maximal diameter) × (perpendicular diameter)2 × 0.52. The results are expressed as the mean of each treatment group ± SD. As shown in Fig. 5a, compared to the Ad-PDCD5 group, significant suppression was observed on days 14–28 in the SG611-PDCD5 groups (P < 0.05 on days 14, 16 and 18; P < 0.01 on days 20, 22, 24, 26 and 28). Compared to the SG611 group, significant suppression was observed on days 24, 26 and 28 in the SG611-PDCD5 groups (all P < 0.01). After excision on day 28, the final tumor sizes in the control, Ad-PDCD5, SG611 and SG611-PDCD5 groups were 2,223.19 ± 916.81, 2,080.75 ± 1,068.27, 1,264.46 ± 545.55 and 371.22 ± 113.15 mm3, respectively. The results suggest that SG611-PDCD5 treatment alone could effectively suppress tumor growth in vivo, and tumor inhibition was superior to SG611 or Ad-PDCD5 treatment alone.

Antitumor activity of SG611-PDCD5 in mice. a Tumors were measured every other day after infection with SG611-PDCD5. The results are expressed as the mean from each treatment group ±SD. Compared to the Ad-PDCD5 group, significant suppression was observed on days 14–28 in the SG611-PDCD5 groups (P < 0.05 on days 14, 16 and day18; P < 0.01 on days 20, 22, 24, 26 and day 28). Compared to the SG611 group, significant suppression was observed on days 24, 26 and day 28 in the SG611-PDCD5 groups (all P < 0.01). After excision on day 28, the final tumor sizes in the control, Ad-PDCD5, SG611 and SG611-PDCD5 groups were 2,223.19.4 ± 916.81, 2,080.75 ± 1,068.27, 1,264.46 ± 545.55 and 371.22 ± 113.15 mm3, respectively. b TUNEL staining to detect apoptotic cells in excised tumors after infection with SG611-PDCD5. Greater numbers of blue–black cells were detected in the SG611-PDCD5 group than in the Ad-PDCD5 or SG611 group
Increased apoptosis observed in tumors treated with SG611-PDCD5
Tumors were excised on day 28. TUNEL staining to detect apoptotic cells in excised tumors. As shown in Fig. 5b, significantly more apoptotic cells were observed in the SG611-PDCD5 group than in SG611 group (19.5 ± 9.50% vs. 6.75 ± 1.50%, P < 0.01). These results suggest that apoptosis is a cytotoxic mechanism occurring in tumors treated with SG611-PDCD5 in vivo.
Discussion
PDCD5 (programmed cell death 5), also designated TF-1 cell apoptosis-related gene-19 (TFAR19), was identified in TF-1 cells undergoing apoptosis [11]. PDCD5 is expressed in lower levels in the heptocellular carcinoma, breast cancer, gastric cancer, astrocytic gliomas and leukemia [15–21], suggesting that reduced PDCD5 expression may play an important role in the mechanism of cancer pathogenesis. We found lower PDCD5 in both AML and CML marrow cells than in normal donor marrow cells. A negative correlation was found between relative levels of PDCD5 and BCR/ABL expression in all CML patients and in CML patients in the advanced phase. Treatment with the ABL tyrosine kinase inhibitor Imatinib mesylate increased PDCD5 expression in K562 and MEG-01 cells. These findings suggest that abnormal expression of PDCD5 in leukemia may be involved in the pathomechanism of AML and CML. Our results showed that exogenous PDCD5 delivered by adenovirus-mediated gene transfer in leukemia cells can markedly enhance sensitivity to the idarubicin in vitro and in vivo [4]. The combined chemogene therapy of IDR and Ad-PDCD5 may lead to development of new strategies for effective leukemia treatment with a potential reduction in systemic toxicity. Future studies will focus on improving transfection rates of PDCD5 in leukemia cells, which may increase the effectiveness and clinical feasibility of the chemogene therapy strategy.
CRAds are the viruses that do not replicate in normal cells but selectively replicate in tumor cells, kill them, and subsequently infect adjacent tumor cells. They hold the promise of being effective agents for the treatment of solid tumors [9, 23, 24]. A few investigations showed that the existence of hypoxic areas in tumor tissues and the expression of telomerase in cancer cells can be employed to achieve cancer targeting treatment [25–31]. Accordingly, the modifications to regulate SG600 with two promoters could restrict adenoviral replication to tumors and thus achieve better tumor-selective oncolysis. SG600 was synchronously deleted 24 nucleotides in E1a-CR2 region that is responsible for binding/inactivating pRb family members, which may partly preserve pRb functions and exert the inhibition of tumor growth. The construction of a gene-viral vector, SG600-p53, by inserting the anticancer gene p53 into the genome of the tumor-specific CRAds can replicate preferably in the cancer cell lines but markedly decreased in normal cell lines [10]. The replicative capability of SG600-p53 ranged from 793.97 to 4,0625.00-fold increase at 96 h after infection, whereas from 1.60- to 50.50-fold increase in normal cells at 96 h after infection. SG600-p53 can increase the expression of p53 and be able to combine the advantages of gene therapy and virotherapy [10]. Recent data in different animal models suggest that SG600-p53 is a safe antitumor therapeutic agent [32].
In the study, SG611-PDCD5, a triple-regulated CRAd carrying PDCD5 gene expression cassette, was constructed based on the SG600 system. The infection efficiency of SG611-EGFP was increased gradually along with the increase of MOI or increased gradually along with time prolonging. SG600-PDCD5 expressed PDCD5 with high efficiency in leukemia cells compared with Ad-PDCD5, showing that the expression of PDCD5 was increased along with the viral replication in leukemia cells. As shown in Fig. 4, the cell viabilities were decreased gradually along with the increase of MOI in these leukemia cells with SG611-PDCD5 in comparison with SG611 and Ad-PDCD5. However, the cell viabilities were maintained a relatively higher level in normal cell lines BJ and L-02 with SG611-PDCD5 even at a MOI of 1,000 and 1,280 pfu/cell, respectively. We further evaluated the antitumor efficacy of SG611-PDCD5 on K562 tumor xenografts established in nude mice. As shown in Fig. 5a, compared to the Ad-PDCD5 group, significant suppression was observed on days 14–28 in the SG611-PDCD5 groups (P < 0.05 on days 14, 16 and 18; P < 0.01 on days 20, 22, 24, 26 and 28). Compared to the SG611 group, significant suppression was observed on days 24, 26 and 28 in the SG611-PDCD5 groups (all P < 0.01). By pathologic examination, we found that the virus administration resulted in leukemia cell apoptosis. The results suggest that SG611-PDCD5 treatment alone could effectively suppress tumor growth in vitro and in vivo, and tumor inhibition was superior to SG611 or Ad-PDCD5 treatment alone.
In conclusion, we successfully constructed an efficient tumor-selective oncolytic adenovirus SG611-PDCD5, which was regulated under both hTERT promoter and HRE and synchronously carried the PDCD5 transgene and E1a-CR2 partially deletion. SG611-PDCD5 holds an increase of anticancer efficacy and might be an improvement of the safety as a new anticancer agent. Future studies will focus on increasing the effectiveness and clinical feasibility of our gene-viral therapy strategy.
References
Miyake H, Hara I, Hara S, Arakawa S, Kamidono S (2000) Synergistic chemosensitization and inhibition of tumor growth and metastasis by adenovirus-mediated P53 gene transfer in human bladder cancer model. Urology 56:332–336
Tuve S, Racek T, Niemetz A, Schultz J, Soengas MS, Putzer BM (2006) Adenovirus-mediated TA-p73beta gene transfer increases chemosensitivity of human malignant melanomas. Apoptosis 11:235–243
Okumura K, Nakase M, Nakamura S, Kamei T, Inui M, Tagawa T (2007) Bax gene therapy for human osteosarcoma using cationic liposomes in vivo. Oncol Rep 17:769–773
Ruan GR, Zhao HS, Chang Y et al (2008) Adenovirus-mediated PDCD5 gene transfer sensitizes K562 cells to apoptosis induced by idarubicin in vitro and in vivo. Apoptosis 13:641–648
Su C, Peng L, Sham J et al (2006) Immune gene-viral therapy with triplex efficacy mediated by oncolytic adenovirus carrying an interferon-gamma gene yields efficient antitumor activity in immunodeficient and immunocompetent mice. Mol Ther 13:918–927
Jounaidi Y, Doloff JC, Waxman DJ (2007) Conditionally replicating adenoviruses for cancer treatment. Curr Cancer Drug Targets 7:285–301
Su CQ, Sham J, Xue HB et al (2004) Potent antitumoral efficacy of a novel replicative adenovirus CNHK300 targeting telomerase-positive cancer cells. J Cancer Res Clin Oncol 130:591–603
Zhang Q, Nie M, Sham J et al (2004) Effective gene-viral therapy for telomerase-positive cancers by selective replicative-competent adenovirus combining with endostatin gene. Cancer Res 64:5390–5397
Zhang Q, Chen G, Peng L et al (2006) Increased safety with preserved antitumoral efficacy on hepatocellular carcinoma with dual-regulated oncolytic adenovirus. Clin Cancer Res 12:6523–6531
Wang X, Su C, Cao H et al (2008) A novel triple-regulated oncolytic adenovirus carrying p53 gene exerts potent antitumor efficacy on common human solid cancers. Mol Cancer Ther 7:1598–1603
Liu H, Wang Y, Zhang Y et al (1999) TFAR19, a novel apoptosis-related gene cloned from human leukemia cell line TF-1, could enhance apoptosis of some tumor cells induced by growth factor withdrawal. Biochem Biophys Res Commun 254:203–210
Wang Y, Li X, Wang L et al (2004) An alternative form of paraptosis-like cell death, triggered by TAJ/TROY and enhanced by PDCD5 overexpression. J Cell Sci 117:1525–1532
Chen Y, Douglass T, Jeffes EW et al (2002) Living T9 glioma cells expressing membrane macrophage colony-stimulating factor produce immediate tumor destruction by polymorphonuclear leukocytes and macrophages via a “paraptosis”-induced pathway that promotes systemic immunity against intracranial T9 gliomas. Blood 100:1373–1380
Dal Canto MC, Gurney ME (1994) Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am J Pathol 145:1271–1279
Xu XR, Huang J, Xu ZG et al (2001) Insight into hepatocellular carcinogenesis at transcriptome level by comparing gene expression profiles of hepatocellular carcinoma with those of corresponding noncancerous liver. Proc Natl Acad Sci USA 98:15089–15094
Hedenfalk I, Duggan D, Chen Y et al (2001) Gene-expression profiles in hereditary breast cancer. N Engl J Med 344:539–548
Zucchi I, Mento E, Kuznetsov VA et al (2004) Gene expression profiles of epithelial cells microscopically isolated from a breast-invasive ductal carcinoma and a nodal metastasis. Proc Natl Acad Sci USA 101:18147–18152
Yang YH, Zhao M, Li WM et al (2006) Expression of programmed cell death 5 gene involves in regulation of apoptosis in gastric tumor cells. Apoptosis 11:993–1001
Li H, Wang Q, Gao F et al (2008) Reduced expression of PDCD5 is associated with high-grade astrocytic gliomas. Oncol Rep 20:573–579
Ruan GR, Qin YZ, Chen SS et al (2006) Abnormal expression of the programmed cell death 5 gene in acute and chronic myeloid leukemia. Leuk Res 30:1159–1165
Ma X, Ruan G, Wang Y et al (2005) Two single-nucleotide polymorphisms with linkage disequilibrium in the human programmed cell death 5 gene 5’ regulatory region affect promoter activity and the susceptibility of chronic myelogenous leukemia in Chinese population. Clin Cancer Res 11:8592–8599
LaBarre DD, Lowy RJ (2001) Improvements in methods for calculating virus titer estimates from TCID50 and plaque assays. J Virol Methods 96:107–126
Yu W, Fang H (2007) Clinical trials with oncolytic adenovirus in China. Curr Cancer Drug Targets 7:141–148
Garber K (2006) China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst 98:298–300
Wirth T, Kuhnel F, Kubicka S (2005) Telomerase-dependent gene therapy. Curr Mol Med 5:243–251
Post DE, Devi NS, Li Z et al (2004) Cancer therapy with a replicating oncolytic adenovirus targeting the hypoxic microenvironment of tumors. Clin Cancer Res 10:8603–8612
Cho WK, Seong YR, Lee YH et al (2004) Oncolytic effects of adenovirus mutant capable of replicating in hypoxic and normoxic regions of solid tumor. Mol Ther 10:938–949
Cogulu O, Kosova B, Karaca E et al (2004) Evaluation of telomerase mRNA (hTERT) in childhood acute leukemia. Leuk Lymphoma 45:2477–2480
Huh HJ, Huh JW, Yoo ES et al (2005) hTERT mRNA levels by real-time RT-PCR in acute myelogenous leukemia. Am J Hematol 79:267–273
Tchirkov A, Chaleteix C, Magnac C et al (2004) hTERT expression and prognosis in B-chronic lymphocytic leukemia. Ann Oncol 15:1476–1480
Bock O, Serinsoz E, Schlue J, Kreipe H (2004) Different expression levels of the telomerase catalytic subunit hTERT in myeloproliferative and myelodysplastic diseases. Leuk Res 28:457–460
Su C, Cao H, Tan S et al (2008) Toxicology profiles of a novel p53-armed replication-competent oncolytic adenovirus in rodents, felids, and nonhuman primates. Toxicol Sci 106:242–250
Acknowledgments
This study was supported by the grant from National Natural Science Foundation of China (No.: 30670894) and supported in part by the grant from the National High Technology Research and Development Program of China (863 Program) (No.: 2006AA02A405).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Xie, M., Niu, JH., Chang, Y. et al. A novel triple-regulated oncolytic adenovirus carrying PDCD5 gene exerts potent antitumor efficacy on common human leukemic cell lines. Apoptosis 14, 1086–1094 (2009). https://doi.org/10.1007/s10495-009-0373-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-009-0373-3