
Abstract
V-set and transmembrane domain-containing 1 (VSTM1), which is downregulated in bone marrow cells from leukemia patients, may provide a diagnostic and treatment target. Here, a triple-regulated oncolytic adenovirus was constructed to carry a VSTM1 gene expression cassette, SG611-VSTM1, and contained the E1a gene with a 24-nucleotide deletion within the CR2 region under control of the human telomerase reverse transcriptase promoter, E1b gene directed by the hypoxia response element, and VSTM1 gene controlled by the cytomegalovirus promoter. Real-time quantitative PCR and Western blot analyses showed that SG611-VSTM1 expressed VSTM1 highly efficiently in the human leukemic cell line K562 compared with SG611. In Cell Counting Kit-8 and flow cytometric assays, SG611-VSTM1 exhibited more potent anti-proliferative and pro-apoptotic effects in leukemic cells compared with SG611 and exerted synergistic cytotoxicity with low-dose daunorubicin (DNR) in vitro. In xenograft models, SG611-VSTM1 intratumorally injected at a dose of 1 × 109 plaque forming units combined with intraperitoneally injected low-dose DNR displayed significantly stronger antitumor effects than either treatment alone. Histopathologic examination revealed that SG611-VSTM1 induced apoptosis of leukemic cells. These results implicate an important role for VSTM1 in the pathogenesis of leukemia, and SG611-VSTM1 may be a promising agent for enhancing chemosensitivity in leukemia therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Benefiting from the development of recombinant DNA technology and molecular-based therapeutics, gene therapy has become an important part of oncotherapy. Immune-related factors, such as GMCSF, CD40L, CD154 and MDA-7/IL-24 [1–4] have been used in gene therapy to directly stimulate cell activation, proliferation and differentiation, induce apoptosis or synergistically enhance the effects of chemotherapy.
V-set and transmembrane domain containing 1 (VSTM1), a potential leukocyte differentiation antigen gene identified by immunogenomic analysis, encodes two main splicing isoforms, VSTM1-v1 and VSTM1-v2 [5, 6]. VSTM1-v1 is a type I membrane molecule and is mainly expressed in human peripheral blood granulocytes and monocytes [7]. VSTM1-v2 is a soluble glycoprotein and can promote the differentiation and activation of Th17 cells [8]. VSTM1 has been reported to be highly expressed in normal myeloid cells, but silenced in multiple leukemic cell lines and downregulated in bone marrow cells from leukemia patients compared with healthy donors [9]. Moreover, restoration of VSTM1-v1 expression in the leukemic cell lines could inhibit cell growth [5, 9], indicating that VSTM1 may be a tumor-specific molecular target and improving its expression in leukemic cells may be useful for leukemia therapy.
Oncolytic adenoviruses, also known as conditionally replicating adenoviruses (CRAds), can not only destroy cancer cells, but also be armed with therapeutic transgenes to generate greater antitumor effects [10]. The genome is large enough to accommodate the incorporation of foreign genes with high transfection efficiency, while the risk of recombination and mutation is low as the viral genome does not integrate into the cell genome [11]. They have been used in different preclinical and clinical studies, showing their capacity to specifically kill tumor cells without major adverse events [12].
Based on two common characteristics of high telomerase activation and low oxygen environment in most cancers [13, 14], Qian et al. previously constructed a mono-regulated CRAd, CNHK300 [15], a dual-regulated CRAd, CNHK500 [16], and a triple-regulated CRAd, SG600 [17], with a deletion of 24 nucleotides within the CR2 region of E1a; meanwhile, the E1a and E1b genes were controlled by the human telomerase reverse transcriptase (hTERT) promoter and the hypoxia response element (HRE), respectively. Remarkably, SG600-p53 and SG611-PDCD5, constructed based on the SG600 system, carrying the p53 or PDCD5 gene expression cassette, displayed more effective tumor-selective killing capacities than replication-defective Ad-p53 and Ad-PDCD5, respectively [17, 18]. In a prior study, we demonstrated that Ad-PDCD5 could sensitize K562 cells to apoptosis induced by etoposide and idarubicin [19, 20], providing a novel chemosensitization strategy for leukemia therapy.
Prompted by features of CRAds and the hypothesis that the VSTM1 gene may play an important biological role in leukemogenesis, in this study we constructed a triple-regulated CRAd carrying the VSTM1 gene expression cassette, SG611-VSTM1. Daunorubicin (DNR) is an effective chemotherapeutic agent widely used in cancer treatment [21], but it is accompanied by severe side effects at increased doses [22, 23]. Here, we examined the impacts of SG611-VSTM1 on the chemosensitivity of leukemic cells to low-dose DNR in vitro and the antitumor effects of SG611-VSTM1 and DNR in vivo. We believe that VSTM1 may represent a therapeutic target for leukemia, and SG611-VSTM1 can be a valuable option for leukemia therapy, particularly in combination with chemotherapeutic drugs.
Materials and methods
Cell culture
The human embryo kidney cell line (HEK293) was purchased from Microbix Biosystems Inc. (Toronto, Ontario, Canada). The human chronic myelogenous leukemic cell line K562, human megakaryoblastic leukemic cell line MEG-01, human acute promyelocytic leukemic cell lines NB4 and HL-60, and human acute lymphoblastic leukemic cell lines SUP-B15 and BV-173 were maintained in RPMI1640 (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) containing 10 % fetal bovine serum (FBS) (Gibco), 100 U/ml penicillin and 100 µg/ml streptomycin (HyClone, Logan, UT, USA). The human fibroblast cell line BJ and human hepatic cell line L-02 were maintained in Dulbecco’s Modified Eagle Medium (DMEM, Gibco) containing 10 % FBS and penicillin/streptomycin. Cells were cultured in a humidified atmosphere containing 5 % CO2 at 37 °C and harvested in the exponential growth phase.
Virus construction
The vector pSG600 mentioned above was kindly provided by the Laboratory of Viral and Gene Therapy, Eastern Hepatobiliary Surgical Hospital (Shanghai, China). The plasmids pClon25 and pPE3-F11B were purchased from Baize Biological Technology Co., Ltd. (Shanghai, China). The VSTM1 cDNA template was kindly provided by the Peking University Human Disease Genome Center (Beijing, China).
First, the PCR-VSTM1 fragment was inserted into pClon25-INS-TK by using NcoI and SalI after amplification and identification. The product was transformed into DH5α Escherichia coli, and positive transformants were isolated by ampicillin selection. The novel vector pClon25-INS-VSTM1 was confirmed by PvuII single digestion and PstI/BglI double digestion. Second, the VSTM1 fragment with a cytomegalovirus promoter and SV40 polyA was released from pClon25-INS-VSTM1 and ligated with pSG600-EGFP by using PacI and NotI-HF. The obtained vector pSG600-VSTM1 was confirmed by PstI and SacI single digestions, as well as HindIII/NcoI and XhoI/KpnI double digestions. Lastly, pPE3-F11B, which is the backbone of the adenovirus with the fiber knob of adenovirus serotype 11B, was co-transfected with pSG600-VSTM1 into HEK293 cells using Lipofectamine 2000 (Gibco) according to the manufacturer’s instructions. The recombinant CRAd, tumor-specific oncolytic adenovirus type 11 carrying the VSTM1 gene, was confirmed by PCR and designated SG611-VSTM1. Similarly, CRAd SG611 was constructed as a blank control.
HEK293 cells were incubated in 10 % FBS/DMEM and infected with SG611-VSTM1 and SG611. Upon replicating to an adequate titer in HEK293 cells, the viruses were purified by a routine method with CsCl2. High performance liquid chromatography (HPLC) was used to determine viral purity. HEK293 cells were seeded in 96-well plates (Corning Costar, Corning, NY, USA) at a density of 1 × 104 per well for 24 h, and subsequently eight serial tenfold dilutions of purified viruses were added to the wells (100 µl/well). Ten replicate wells were used for each dilution (10−6–10−13), and two wells were used as negative control. The cytopathic effect was detected after co-culture for 10 days. The virus titer was determined with the 50 % tissue culture infectious doses (TCID50) method [24].
RQ-PCR
K562 cells were plated in 6-well plates at a density of 8 × 105 per well and exposed to SG611-VSTM1 and SG611 at the multiplicity of infection (MOI) of 1 and 5 plaque-forming units (pfu) per cell, with normal saline as a control. Each group was tested in triplicate. On days 0, 1, 2, 3, 4, 5 and 6 after infection, the cells were collected, and the cellular RNA was extracted using the TRIzol®reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The cDNA synthesis reaction was performed and was used to detect the VSTM1 expression level by real-time quantitative PCR (RQ-PCR) on the ABI7500 Sequence Detection System (Applied Biosystems, Carlsbad, CA, USA) as described previously [25]. The Abelson (ABL) gene was used as an internal standard.
Western blot
Similarly, K562 cells were infected with SG611-VSTM1 and SG611 at the MOI of 1 and 5 pfu/cell. On days 1, 3, and 5 after infection, the cellular proteins were extracted, and protein concentrations were determined using the BCA Protein Assay Kit (Pierce, Rockford, IL, USA) as described by the manufacturer. Protein lysates were separated on 10 % SDS–polyacrylamide gels and electro-transferred to nitrocellulose membranes (Millipore, Bedford, MA, US). Western blotting was performed as previously described [5]. β-actin was used as an internal standard to indicate the amount of input lysates and verify the integrity of these lysates.
CCK-8 assay
Cell viability was assessed by the Cell Counting Kit-8 (CCK-8) assay. K562, MEG-01, NB4, SUP-B15, BV-173, HL-60, L-02 and BJ cells were seeded in 96-well plates at a density of 1 × 104 per well and infected with SG611-VSTM1 or SG611, with normal saline as a control, at increasing MOI from 0 to 1280 pfu/cell. To evaluate the cytotoxicity of SG611-VSTM1 in combination with DNR, cell lines K562, MEG-01, NB4 and SUP-B15 were treated with 40, 120 or 240 ng/ml DNR (Cayman Chemical Co., Ann Arbor, MI, USA) for an additional 24 h after infection with SG611-VSTM1 for 48 h. Each group was provided with three wells. The CCK-8 assay was separately performed at 72, 96, 120 and 168 h after infection. CCK-8 agent (10 µl, Dojindo, Japan) was added to each well, and then the cells were incubated for an additional 1–2 h. Absorbance was read at 450 nm on a scanning multiwell spectrophotometer with a reference wavelength at 570 nm (Bio-Rad 650, Hercules, CA, USA).
Flow cytometry
K562 and MEG-01 cells were cultured in 24-well plates at a density of 1 × 105 per well and infected with SG611-VSTM1 and SG611 at the MOI of 10, 40, 80 and 160 pfu/cell for 48 and 72 h. To study the combined effect of SG611-VSTM1 and DNR on cell apoptosis, the cells were infected with SG611-VSTM1, followed 24 h later by treatment with 40, 120 and 240 ng/ml DNR for an additional 24 h. The cells were collected, washed with phosphate buffered saline (PBS, Solarbio, Beijing, China) and Annexin V Binding Buffer (Becton–Dickinson, San Jose, CA, USA) successively, and resuspended in 100 μl binding buffer. PE Annexin V (5 μl) was added as well as 7-AAD (5 μl) according to the protocol of the Apoptosis Detection Kit I (Becton–Dickinson). After incubation for 15 min at room temperature in the dark, 100 μl Binding Buffer was added, and samples were immediately analyzed on a FACSCalibur flow cytometer (Becton–Dickinson). Apoptosis could be measured over time by tracking the change in labeling of cells from PE Annexin V-/7-AAD- (viable), to PE Annexin V+/7-AAD-(early apoptosis) and finally to PE Annexin V+/7-AAD + (end stage apoptosis and death). Each analysis was performed using at least 20,000 events.
Animal experiments
BALB/c nu/nu female mice (age 4–5 weeks; weighing 15–16 g) at the Experimental Animal Center of Peking University People’s Hospital (Beijing, China) were used in this study. All care and handling of animals were performed with the approval of Institutional Authority for Laboratory Animal Care of Peking University People’s Hospital. Mice were pretreated with 100 mg/kg cyclophosphamide by intraperitoneal (i.p.) injection once daily for 2 consecutive days. Two days later, 1.0 × 107 K562 cells in a total volume of 100 μl were subcutaneously (s.c.) injected into the right flank of each mouse. When the diameters of the tumors reached 6–8 mm, mice were randomly divided into five groups, as follows: six mice for Group I, SG611; seven mice for Group II, SG611-VSTM1; seven mice for Group III, DNR; eight mice for Group IV, SG611-VSTM1 plus DNR and six mice for Group V, with normal saline as a control. Mice in Groups I, II and IV were given a total of 1 × 109 pfu viruses by intratumoral injection administered once every other day for 5 times. Starting from the next day following the first injection of viruses, mice in Groups III and IV were i.p. treated with 300 μg/kg DNR once every other day, for a total of five times. In Group V, normal saline (50 μl per mouse each time) was injected intratumorally or i.p. Tumor sizes were measured every day. Tumor volume was calculated using the following formula: (maximal diameter) × (perpendicular diameter)2 × 0.5 [26]. Results were expressed as the mean of each group ± standard deviation (SD).
TUNEL assay
Mice were sacrificed, and tumors were excised on day 13. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining (Roche, Basel, Switzerland) was used to detect apoptosis in formalin-fixed, paraffin-embedded tumor tissues. The samples were digested with proteinase K at 37 °C for 30 min and then incubated with the TUNEL reaction mixture containing terminal deoxynucleotidyl transferase and fluorescein-dUTP at 37 °C for 1 h in a humidified atmosphere in the dark. The slides were washed twice and incubated with converter-POD at 37 °C for 30 min. Sections were visualized with 3,3′-diaminobenzidine (DAKO, Glostrup, Denmark) using a light microscope, and nuclei appearing brown were considered positive cells. At 400× magnification, the eyepiece provided a field size of 0.24 mm2. Results were expressed as the mean of apoptotic cells per field ± SD, and three different fields per slide were examined.
Statistical analysis
Statistical analyses were performed using SPSS 20.0 (Chicago, IL, USA). Data from in vitro experiments were assessed by Student’s t test, and data from in vivo experiments were assessed by one-way analysis of variance (ANOVA). Experimental figures were generated by using GraphPad Prism 5.0 (San Diego, CA, USA). P < 0.05 was considered statistically significant.
Results
Generation of a recombinant triple-regulated oncolytic adenovirus, SG611-VSTM1
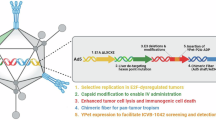
The novel triple-regulated oncolytic adenovirus carrying VSTM1 gene expression cassette, SG611-VSTM1, was constructed and confirmed. In SG611-VSTM1, the E1a gene with a deletion of 24 nucleotides within the CR2 region was controlled under the hTERT promoter, the E1b gene was directed by the HRE, whereas the VSTM1 gene was controlled by the cytomegalovirus promoter (Fig. 1). HPLC showed that the viral purity was over 98 %. The titers of SG611-VSTM1 and SG611 were 5.71 × 1010 and 2.06 × 1010 TCID50/ml, respectively.

Diagram of SG611-VSTM1 construct. A VSTM1 gene expression cassette containing VSTM1 cDNA, cytomegalovirus promoter and SV40 polyA was inserted into the genome of SG611 to generate SG611-VSTM1. ITR, inverted terminal repeats; ψ, adenovirus type 5 packaging signal
SG611-VSTM1 expresses VSTM1 with high efficiency in leukemic cell line K562
To investigate the infection efficiency of SG611-VSTM1 and the relative VSTM1 expression levels in leukemic cells, RQ-PCR and Western blot were performed. RQ-PCR analysis (Fig. 2a) showed that VSTM1 was not expressed in untreated K562 cells, K562 cells treated with normal saline or K562 cells infected with SG611. By contrast, the expression level of VSTM1 in K562 cells infected with SG611-VSTM1 increased gradually along with the increase of MOI and time after infection. At the MOI of 5 pfu/cell, the ratios of VSTM1/ABL on days 1, 2, 3, 4, 5 and 6 were 0.11 ± 0.01, 1.52 ± 0.25, 5.85 ± 0.91, 36.08 ± 24.86, 70.24 ± 16.20 and 240.52 ± 34.13, respectively. SG611-VSTM1 expressed VSTM1 in K562 cells efficiently compared with SG611 (P < 0. 001). Similar results were observed in Western blot assays (Fig. 2b).

VSTM1 expression in K562 cells after infection with SG611-VSTM1 and SG611. a RQ-PCR analysis showed that SG611-VSTM1 expressed VSTM1 in K562 more efficiently than SG611. The expression level of VSTM1 increased along with the increase in MOI of SG611-VSTM1 and the length of time after infection. b Western blot assays showed similar results
SG611-VSTM1 selectively inhibits leukemic cell proliferation in vitro
Next, we evaluated the effect of SG611-VSTM1 on cell proliferation using six human leukemic cell lines and two normal cell lines by the CCK-8 assay. As shown in Fig. 3, the viabilities of these leukemic cells decreased gradually along with the increase of MOI. Compared with SG611, SG611-VSTM1 displayed a more potent inhibitory effect on leukemic cell proliferation. At 96 h after infection at the MOI of 80, 120 and 160 pfu/cell, the viability of K562 cells infected with SG611-VSTM1 was lower than that of cells infected with SG611 at the corresponding dose (all P < 0.01). Similar results were observed in the following cell lines: MEG-01 at 72 h after infection at the MOI of 40, 80, 160 and 320 pfu/cell; HL-60 at 72 h after infection at the MOI of 40, 80, 160 and 320 pfu/cell; BV-173 at 96 h after infection at the MOI of 80, 120 and 160 pfu/cell; NB4 at 96 h after infection at the MOI of 80, 120 and 160 pfu/cell; SUP-B15 at 120 h after infection at the MOI of 80, 120 and 160 pfu/cell (all P < 0.01). However, the cell viabilities remained at a relatively higher level in the normal cell lines BJ (about 50 %) and L-02 (about 60 %) after infection with SG611-VSTM1 at the MOI of 1280 pfu/cell.

Cell viability of leukemic cell lines and normal cell lines after infection with SG611-VSTM1 and SG611. The viabilities of these leukemic cells decreased gradually along with the increase of MOI, especially in the SG611-VSTM1 group. However, the cell viabilities remained at a relatively higher level in the normal cell lines BJ (about 50 %) and L-02 (about 60 %) after infection with SG611-VSTM1 at the MOI of 1280 pfu/cell. *represents P < 0.05, **represents P < 0.01
SG611-VSTM1 effectively promotes leukemic cell apoptosis in vitro
Using PE Annexin V/7-AAD staining, we assessed if SG611-VSTM1 could influence the apoptosis of leukemic cells. In K562 cells, a significant increase in the percentage of apoptotic cells was observed, especially early apoptotic cells, along with the increases of MOI and time post-infection (Fig. 4). At 48 h after infection at the MOI of 40 and 160 pfu/cell, the percentages of apoptotic cells exposed to SG611-VSTM1 were significantly higher at 19.3 and 70.9 % compared with those infected with SG611 (11.0 and 46.6 %), respectively. Similar results were found at 72 h after infection. In MEG-01 cells, similar results also showed that the pro-apoptotic effect on leukemic cells of SG611-VSTM1 was superior to that of SG611.

Apoptosis of K562 and MEG-01 cells after infection with SG611-VSTM1 and SG611. The percentage of apoptotic cells increased gradually along with the increase of MOI and length of time post-infection, especially in the SG611-VSTM1 group
SG611-VSTM1 and DNR synergistically inhibit leukemic cell proliferation
To examine the effect of VSTM1 overexpression on the cellular response to combination treatment with DNR, K562, NB4, MEG-01 and SUP-B15 cells were infected with SG611-VSTM1, followed 48 h later with DNR treatment for 24 h. We found that the viabilities of these leukemic cells were obviously decreased after infection with SG611-VSTM1 alone or treatment with DNR alone (Fig. 5). In K562 cells, the cell viability (%) of SG611-VSTM1 at the MOI of 80 pfu/cell in combination with 120 ng/ml DNR was 29.07 ± 1.38, significantly lower than that of SG611-VSTM1 alone (38.64 ± 5.58, P = 0. 017) or DNR alone (90.79 ± 3.39, P < 0. 001). Similar trends were seen in NB4, MEG-01 and SUP-B15 cells, indicating that SG611-VSTM1 and DNR synergistically inhibited leukemic cell proliferation.

Cell viability of leukemic cell lines after infection with SG611-VSTM1 followed by DNR treatment. Compared with SG611-VSTM1 and DNR treatment alone, the combined treatment exerted a stronger inhibitory effect on leukemic cell proliferation
SG611-VSTM1 and DNR synergistically promote leukemic cell apoptosis
K562 and MEG-01 cells were infected with SG611-VSTM1, followed 24 h later by treatment with low-dose DNR for 24 h. As shown in Fig. 6, in K562 cells, the percentage of apoptotic cells was not obviously changed after treatment with 40 ng/ml DNR compared with the control (8.3 vs. 3.2 %), but it was significantly different after treatment with 120 ng/ml DNR (13.5 vs. 3.2 %). Compared with the single treatment group, an increased percentage of apoptotic cells was observed in the combined treatment group. The percentage of apoptotic cells infected with SG611-VSTM1 at the MOI of 80 pfu/cell in combination with 120 ng/ml DNR was 51.7 %, significantly higher than that of SG611-VSTM1 alone (32.1 %) or DNR alone (13.5 %). The results also showed that SG611-VSTM1 combined with DNR enhanced apoptosis in a dose-dependent manner (27.8 vs. 39.5 %, combined with 40 ng/ml DNR; 30.7 vs. 51.7 %, combined with 120 ng/ml DNR). Similar results were observed in MEG-01 cells.

Apoptosis of K562 and MEG-01 cells after infection with SG611-VSTM1 followed by DNR treatment. The percentage of apoptotic cells in the combined treatment group was significantly higher than those in the SG611-VSTM1 group and DNR group
SG611-VSTM1 and DNR exert potent antitumor effects in vivo
Xenograft K562 tumor models in nude mice were used to evaluate the antitumor effects of SG611-VSTM1 in vivo. As shown in Fig. 7a, compared with the control group, significant suppression was observed in the SG611-VSTM1 group on days 3–13 (P < 0.05 on day 3; P < 0.01 on days 4–13), in the SG611 group on days 3–13 (P < 0.05 on days 3 and 5; P < 0.01 on days 4 and 6–13), and in the DNR group on days 7 and 9–13 (all P < 0.05). Remarkably, the combined treatment resulted in more significant tumor suppression on days 9–13 compared with SG611-VSTM1 alone (P = 0.003, P = 0.046, P = 0.047, P = 0.048, P = 0.036, respectively) and DNR alone (all P < 0.01). Tumors were excised on day 13 (Fig. 7b). Accordingly, SG611-VSTM1 alone could completely inhibit tumor growth, and SG611-VSTM1 combined with low-dose DNR exhibited synergistic antitumor activity in vivo.

Antitumor efficacy of SG611-VSTM1 in xenograft K562 tumor models in nude mice. a Tumors were measured every day after infection with viruses. Compared with the control group, significant suppression was observed in the SG611-VSTM1 group on days 3–13 (P < 0.05 on day 3; P < 0.01 on days 4–13), in the SG611 group on days 3–13 (P < 0.05 on days 3 and 5; P < 0.01 on days 4 and 6–13), and in the DNR group on days 7 and 9–13 (all P < 0.05). Remarkably, the combined treatment resulted in more significant tumor suppression on days 9–13 compared with SG611-VSTM1 alone (P = 0.003, P = 0.046, P = 0.047, P = 0.048, P = 0.036, respectively) and DNR alone (all P < 0.01). b Excised tumors on day 13. *A tumor in the SG611 group was too small to be excised
SG611-VSTM1 and DNR kill tumor cells by inducing apoptosis in vivo
Apoptosis in excised tumors were detected by the TUNEL assay. As shown in Fig. 8, the apoptotic cells per field in the control, SG611, SG611-VSTM1, DNR and SG611-VSTM1 plus DNR group were 3.7 ± 0.6, 10.7 ± 3.5, 11.0 ± 2.7, 7.3 ± 2.3, and 20.3 ± 5.1, respectively. More apoptotic cells per field were observed in the SG611-VSTM1 plus DNR group than in the SG611-VSTM1 or DNR group (P = 0.049, P = 0.016, respectively). These results suggest that inducing cell apoptosis is a potential antitumor mechanism of the SG611-VSTM1 and DNR combination in vivo.

Detection of apoptosis in excised tumors after different treatments by TUNEL assay. The apoptotic cells per field in the control, SG611, SG611-VSTM1, DNR and SG611-VSTM1 plus DNR group were 3.7 ± 0.6, 10.7 ± 3.5, 11.0 ± 2.7, 7.3 ± 2.3, and 20.3 ± 5.1, respectively. More apoptotic cells per field were observed in the SG611-VSTM1 plus DNR group than in the SG611-VSTM1 or DNR group (P = 0.049, P = 0.016, respectively)
Discussion
VSTM1, located on chromosome 19q13.42, is a potential leukocyte differentiation antigen gene identified by Peking University Human Disease Genome Center [8]. It has at least five variants, VSTM1-v1 to -v5, among which VSTM1-v1 and VSTM1-v2 (GenBank accession numbers: NM_198481, BC100943) are the functionally predominant forms. VSTM1-v1, also known as signal inhibitory receptor on leukocytes-1 (SIRL-1), is an ITIM-bearing immune receptor that negatively regulates neutrophil activity, with an extracellular IgV-like domain and two ITIM motifs in its cytoplasmic tail [7, 27, 28]. Lacking the transmembrane domain compared with VSTM1-v1, VSTM1-v2 is a classic secreting glycoprotein capable of promoting CD4+ T cells to secret IL-17A [8], and its secretion capacity can be enhanced by the mouse IgGκ signal peptide [29]. Previous studies have identified that VSTM1 is widely expressed in normal human peripheral blood leukocytes, including granulocytes, monocytes and lymphocytes, but undetectable in numerous hematopoietic tumor cell lines with promoter hypermethylation and downregulated in bone marrow cells from leukemia patients compared with healthy donors. Moreover, the expression level is positively correlated with the myeloid cell maturation state [5, 9]. Similar to its function in Jurkat cells [5], plasmid-mediated VSTM1 overexpression in leukemic cell lines K562 and MEG-01 also inhibits cell growth [9]. These results suggest that VSTM1 may play an important role in leukemogenesis.
CRAds are designed to infect, replicate and kill cancer cells while sparing normal cells, and they are expected to be effective vectors for gene therapy for some solid tumors [30, 31]. Two types of genetic modifications lead to two different subclasses of CRAds, type I and type II [12]. Unlike the latter in which the genome is placed under the control of a tumor-specific promoter, the former is characterized by mutations or deletions in the E1 region of the genome, interfering with the inactivation of cell cycle regulators such as p53 and retinoblastoma protein (Rb). For example, SG600 contains a deletion of 24 nucleotides in the E1a-CR2 region, which abrogates E1a-Rb interaction and causes it to replicate mainly in actively dividing tumor cells [12, 32]. Recently, the tumor-selective killing capacities of SG600-p53 and SG611-PDCD5, constructed based on the SG600 system, were demonstrated in several tumor cell lines in vitro and in xenograft models of human non-small-cell lung cancer or leukemic cell line in nude mice [17, 18, 33, 34]. Specifically, CRAds have provided favorable safety profiles for patients in early clinical trials and showed the ability to eradicate cancer stem cells directly through shared targets or indirectly via synergistic effects with chemotherapy and radiation therapy [35], favoring multimodal approaches to cancer therapy.
DNR, an anthracycline chemotherapeutic agent, acting through inducing DNA damage, is commonly used in the treatment of hematopoietic malignancies [21]. However, it can dose-dependently lead to severe myelosuppression and cardiac toxicity [22, 23]. A chemo-gene-viro-therapeutic strategy has emerged as a viable alternative to conventional chemotherapy combinations [36–38]. It works through enhancing the sensitivity of tumor cells to chemotherapeutic drugs at lower doses by utilizing the tumor-selective replication vector carrying a specific gene, thus reducing systemic toxicity. We have reported that adenovirus-mediated overexpression of the PDCD5 gene sensitized K562 cells to apoptosis induced by low-dose anthracycline idarubicin in vitro and in vivo [19], indicating that this strategy may be used in the treatment of leukemia.
In this study, a tumor-selective oncolytic adenovirus carrying the VSTM1 gene expression cassette, SG611-VSTM1, was constructed based on the SG600 system. Our results showed that SG611-VSTM1 expressed VSTM1 with high efficiency in leukemic cells and exhibited more potent anti-proliferative and pro-apoptotic effects on leukemic cells compared with SG611 in vitro. However, the inhibitory effect on normal cell growth was weaker, indicating that SG611-VSTM1 selectively exerted high levels of toxicity in leukemic cells. Similar to the results in vitro, intratumoral injection of SG611-VSTM1 in combination with the i.p. injection of low-dose DNR resulted in synergistic antitumor effects in xenograft models. These findings suggest that the VSTM1 gene may be involved in leukemic cell proliferation and apoptosis. However, no significant difference in tumor size was observed between the SG611-VSTM1 group and the SG611 group in vivo, which may have resulted from the use of excess viruses in this experiment. Thus, the function and the exact mechanism of the VSTM1 gene in vitro and in vivo will need to be further explored.
In conclusion, we successfully constructed a triple-regulated oncolytic adenovirus SG611-VSTM1 with selective cytotoxicity to human leukemic cells. The combination treatment of SG611-VSTM1 and DNR exerted synergistic suppressive effects on leukemic cell growth in vitro and in vivo. This study provides evidence that when leukemic cells are treated with SG611-VSTM1, low doses of DNR can efficiently induce apoptosis. The combined chemogene therapy of DNR and SG611-VSTM1 may lead to the development of new strategies for effective leukemia treatment with a potential reduction in systemic toxicity. Future studies will focus on increasing the effectiveness and clinical feasibility of our gene-viral therapy strategy.
References
Hemminki O, Parviainen S, Juhila J, Turkki R, Linder N, Lundin J et al (2015) Immunological data from cancer patients treated with Ad5/3-E2F-Δ24-GMCSF suggests utility for tumor immunotherapy. Oncotarget 6:4467–4481
Liljenfeldt L, Yu D, Chen L, Essand M, Mangsbo SM (2014) A hexon and fiber-modified adenovirus expressing CD40L improves the antigen presentation capacity of dendritic cells. J Immunother 37:155–162
Castro JE, Melo-Cardenas J, Urquiza M, Barajas-Gamboa JS, Pakbaz RS, Kipps TJ (2012) Gene immunotherapy of chronic lymphocytic leukemia: a phase I study of intranodally injected adenovirus expressing a chimeric CD154 molecule. Cancer Res 72:2937–2948
Ma G, Kawamura K, Shan Y, Okamoto S, Li Q, Namba M et al (2014) Combination of adenoviruses expressing melanoma differentiation-associated gene-7 and chemotherapeutic agents produces enhanced cytotoxicity on esophageal carcinoma. Cancer Gene Ther 21:31–37
Li T, Guo X, Wang W, Mo X, Wang P, Han W (2015) V-set and transmembrane domain-containing 1 is silenced in human hematopoietic malignancy cell lines with promoter methylation and has inhibitory effects on cell growth. Mol Med Rep 11:1344–1351
Li T, Wang W, Chen Y, Han W (2013) Preparation and characterization of monoclonal antibodies against VSTM1. Monoclon Antib Immunodiagn Immunother 32:283–289
Steevels TA, Lebbink RJ, Westerlaken GH, Coffer PJ, Meyaard L (2010) Signal inhibitory receptor on leukocytes-1 is a novel functional inhibitory immune receptor expressed on human phagocytes. J Immunol 184:4741–4748
Guo X, Zhang Y, Wang P, Li T, Fu W, Mo X et al (2012) VSTM1-v2, a novel soluble glycoprotein, promotes the differentiation and activation of Th17 cells. Cell Immunol 278:136–142
Xie M, Li T, Li N, Li J, Yao Q, Han W et al (2015) VSTM-v1 is a potential myeloid differentiation antigen gene that is downregulated in bone marrow cells from patients with myeloid leukemia. J Hematol Oncol 8:25
Jiang G, Xin Y, Zheng JN, Liu YQ (2011) Combining conditionally replicating adenovirus-mediated gene therapy with chemotherapy: a novel antitumor approach. Int J Cancer 129:263–274
Chu RL, Post DE, Khuri FR, Van Meir EG (2004) Use of replicating oncolytic adenoviruses in combination therapy for cancer. Clin Cancer Res 10:5299–5312
Bressy C, Benihoud K (2014) Association of oncolytic adenoviruses with chemotherapies: an overview and future directions. Biochem Pharmacol 90:97–106
Shay JW, Wright WE (2001) Telomeres and telomerase: implications for cancer and aging. Radiat Res 155:188–193
Hewitson KS, Schofield CJ (2004) The HIF pathway as a therapeutic target. Drug Discov Today 9:704–711
Su CQ, Sham J, Xue HB, Wang XH, Chua D, Cui ZF et al (2004) Potent antitumoral efficacy of a novel replicative adenovirus CNHK300 targeting telomerase-positive cancer cells. J Cancer Res Clin Oncol 130:591–603
Zhang Q, Chen G, Peng L, Wang X, Yang Y, Liu C et al (2006) Increased safety with preserved antitumoral efficacy on hepatocellular carcinoma with dual-regulated oncolytic adenovirus. Clin Cancer Res 12:6523–6531
Wang X, Su C, Cao H, Li K, Chen J, Jiang L et al (2008) A novel triple-regulated oncolytic adenovirus carrying p53 gene exerts potent antitumor efficacy on common human solid cancers. Mol Cancer Ther 7:1598–1603
Xie M, Niu JH, Chang Y, Qian QJ, Wu HP, Li LF et al (2009) A novel triple-regulated oncolytic adenovirus carrying PDCD5 gene exerts potent antitumor efficacy on common human leukemic cell lines. Apoptosis 14:1086–1094
Ruan GR, Zhao HS, Chang Y, Li JL, Qin YZ, Liu YR et al (2008) Adenovirus-mediated PDCD5 gene transfer sensitizes K562 cells to apoptosis induced by idarubicin in vitro and in vivo. Apoptosis 13:641–648
Ruan GR, Chen SS, Chang Y, Li JL, Qin YZ, Li LD et al (2007) Adenovirus-mediated PDCD5 gene transfer sensitizes apoptosis of K562 cells induced by etoposide. Zhongguo Shi Yan Xue Ye Xue Za Zhi 15:936–940
Davis H, Davis T (1979) Daunorubicin and adriamycin in cancer treatment: an analysis of their roles and limitations. Cancer Treat Rep 63:809–815
Young R, Ozols R, Myers C (1981) The anthracycline antineoplastic drugs. N Engl J Med 305:139–153
Shan K, Lincoff M, Young J (1996) Anthracycline-induced cardiotoxicity. Ann Intern Med 125:47–58
LaBarre DD, Lowy RJ (2001) Improvements in methods for calculating virus titer estimates from TCID50 and plaque assays. J Virol Methods 96:107–126
Ruan GR, Qin YZ, Chen SS, Li JL, Ma X, Chang Y (2006) Abnormal expression of the programmed cell death 5 gene in acute and chronic myeloid leukemia. Leuk Res 30:1159–1165
Euhus DM, Hudd C, LaRegina MC, Johnson FE (1986) Tumor measurement in the nude mouse. J Surg Oncol 31:229–234
Steevels TA, van Avondt K, Westerlaken GH, Stalpers F, Walk J, Bont L et al (2013) Signal inhibitory receptor on leukocytes-1 (SIRL-1) negatively regulates the oxidative burst in human phagocytes. Eur J Immunol 43:1297–1308
Van Avondt K, Fritsch-Stork R, Derksen RH, Meyaard L (2013) Ligation of signal inhibitory receptor on leukocytes-1 suppresses the release of neutrophil extracellular traps in systemic lupus erythematosus. PLoS One 8:e78459
Liu H, Zou X, Li T, Wang X, Yuan W, Chen Y et al (2016) Enhanced production of secretory glycoprotein VSTM1-v2 with mouse IgGκ signal peptide in optimized HEK293F transient transfection. J Biosci Bioeng 121:133–139
Mu H, Wang N, Zhao L, Li S, Li Q, Chen L et al (2015) Methylation of PLCD1 and adenovirus-mediated PLCD1 overexpression elicits a gene therapy effect on human breast cancer. Exp Cell Res 332:179–189
Yang Y, Xu H, Huang W, Ding M, Xiao J, Yang D et al (2015) Targeting lung cancer stem-like cells with TRAIL gene armed oncolytic adenovirus. J Cell Mol Med 19:915–923
Yamamoto M, Curiel DT (2010) Current issues and future directions of oncolytic adenoviruses. Mol Ther 18:243–250
Chen GX, Zhang S, He XH, Liu SY, Ma C, Zou XP (2014) Clinical utility of recombinant adenoviral human p53 gene therapy: current perspectives. Onco Targets Ther 7:1901–1909
Tazawa H, Kagawa S, Fujiwara T (2013) Advances in adenovirus-mediated p53 cancer gene therapy. Expert Opin Biol Ther 13:1569–1583
Short JJ, Curiel DT (2009) Oncolytic adenoviruses targeted to cancer stem cells. Mol Cancer Ther 8:2096–2102
Pan QW, Zhong SY, Liu BS, Liu J, Cai R, Wang YG et al (2007) Enhanced sensitivity of hepatocellular carcinoma cells to chemotherapy with a Smac-armed oncolytic adenovirus. Acta Pharmacol Sin 28:1996–2004
Wu YM, Zhang KJ, Yue XT, Wang YQ, Yang Y, Li GC et al (2009) Enhancement of tumor cell death by combining cisplatin with an oncolytic adenovirus carrying MDA-7/IL-24. Acta Pharmacol Sin 30:467–477
Ma B, Wang Y, Zhou X, Huang P, Zhang R, Liu T et al (2015) Synergistic suppression effect on tumor growth of hepatocellular carcinoma by combining oncolytic adenovirus carrying XAF1 with cisplatin. J Cancer Res Clin Oncol 141:419–429
Acknowledgments
This study was supported by grants from the National Basic Research Program of China (Grant 2013CB733701), the Specialized Research Fund for the Doctoral Program of Higher Education of China (Grant 20130001110079), the National Natural Science Foundation of China (Grant 81570182), and the Key Program of National Natural Science Foundation of China (Grant 81530046).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Zhou, J., Yao, QM., Li, JL. et al. Synergistic antitumor activity of triple-regulated oncolytic adenovirus with VSTM1 and daunorubicin in leukemic cells. Apoptosis 21, 1179–1190 (2016). https://doi.org/10.1007/s10495-016-1276-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-016-1276-8