
Abstract
A universal cellular defense mechanism against viral invasion is the elimination of infected cells through apoptotic cell death. To counteract host defenses many viruses have evolved complex apoptosis evasion strategies. The oncogenic human retrovirus HTLV-1 is the etiological agent of adult-T-cell leukemia/lymphoma (ATLL) and the neurodegenerative disease known as HTLV-associated myelopathy/tropical spastic paraparesis (HAM/TSP). The poor prognosis in HTLV-1-induced ATLL is linked to the resistance of neoplastic T cells against conventional therapies and the immuno-compromised state of patients. Nevertheless, several studies have shown that the apoptotic pathway is largely intact and can be reactivated in ATLL tumor cells to induce specific killing. A better understanding of the molecular mechanisms employed by HTLV-1 to counteract cellular death pathways remains an important challenge for future therapies and the treatment of HTLV-1-associated diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Apoptosis, or programmed cell death, plays a major role in tissue development, homeostasis, and the immune response [1]. Virus-infected cells are frequently removed from the body through apoptosis, effectively eliminating the infection in the absence of an inflammatory response. Apoptosis is tightly controlled by a group of cysteine proteases known as caspases, as well as the Bcl-2 family of proteins which regulate the release of pro-apoptotic proteins from the mitochondria. Despite multiple levels of regulation, deregulated apoptosis contributes to the development of cancer, while excessive apoptosis is conversely associated with tissue destruction seen in various auto-immune disorders [2]. To regulate apoptosis induced by the host, many viruses have evolved strategies to modulate key checkpoints of the apoptotic pathway. Some viruses, such as members of the γ-herpesvirus family, encode a homologue of cellular anti-apoptotic Bcl-2 [3]. A variety of other novel viral anti-apoptotic mechanisms have been characterized, including: caspase inhibitors (i.e. poxviruses, murine herpes virus-68, and African swine fever virus); soluble cytokine receptors (EBV); the inhibition of cellular stress responses (Papillomaviridae, Polyomaviridae, and Adenoviridae); and the inhibition of death receptor-mediated apoptosis (γ-herpesviruses and poxviruses) [4–7]. A number of DNA viruses, such as poxviruses, adenoviruses, and human cytomegalovirus (CMV), also encode mitochondrial-localized inhibitors of apoptosis which function to regulate cytochrome c release [5].
In stark contrast to these anti-apoptotic mechanisms, other viruses appear to sensitize cells to apoptosis to the benefit of virus replication and egress. Human immunodeficiency virus (HIV) and hepatitis B (HBV) virus encode pro-apoptotic alpha-helical proteins Vpr and HBX that form pores in the mitochondrial membrane [5], thereby sensitizing the mitochondria to cytochrome c release. Other viral proteins, including E1A from adenovirus, the envelope protein from HIV, human papilloma virus (HPV) protein E1E4, the fusion protein from respiratory syncytial virus (RSV), and the reovirus protein mu1, also induce apoptosis through various mechanisms, including the disruption of the mitochondrial network and p53 activation [8, 9]. While some of these viral proteins, such as Vpr and HBX, specifically induce programmed cell death to the benefit of the virus, other viral proteins such as E1A appear to induce apoptosis as a consequence of detection by innate cellular defense mechanisms.
HTLV-1: human T-cell leukemia virus type 1
The retrovirus human T-cell leukemia virus (HTLV)-1 is the etiological agent of adult T-cell leukemia/lymphoma (ATLL), a fatal lymphoproliferative disease [10]. While the majority of HTLV-1-infected individuals remain asymptomatic, upwards of 5% of patients ultimately develop ATLL. ATLL is characterized by the rapid and uncontrolled clonal proliferation of mature transformed CD25+/CD4+ T cells, and the mean survival of patients in the acute phase of the disease is approximately 6 months [11]. HTLV-1-infection is also associated with a neurodegenerative disease known as HTLV-associated myelopathy/tropical spastic paraparesis (HAM/TSP) [12, 13]. Other autoimmune diseases, including uveitis, arthritis, polymyositis, Sjögren syndrome, atopic dermatitis, and alveolitis, have been reported in HTLV-1 infected individuals [13]. Altogether, the treatment of HTLV-1-infected patients is generally difficult as infected cells are refractory to conventional chemotherapy and radiation-based cancer treatments.
HTLV-1-infected cells and ATLL cells from patients are highly resistant to multiple pro-apoptotic stimuli, including death receptor-mediated, DNA damage-induced, and γ-irradiation apoptosis compared to uninfected normal cells [14–18]. HTLV-1-infected ATLL cells removed from the in vivo environment, however, die spontaneously by apoptosis when cultured in vitro, thereby complicating investigations into mechanisms employed by patient-derived ATLL cells [19]. As a result, most studies with ATLL and HTLV-1-infected cells rely on HTLV-1-transformed cells in vitro or short term culture of ATLL derived cells.
In contrast to ATLL, TSP/HAM is associated with chronic and progressive inflammation of the spinal cord [12]. TSP/HAM derived cell lines, like ATLL [20, 21], also exhibit resistance to FasL- and etoposide-induced apoptosis [22, 23], and FasL and the Fas-associated phosphatase are up-regulated in TSP/HAM cells [22, 24, 25]. While TSP/HAM cell lines exhibit a general resistance to apoptosis, expression of the viral protein Tax sensitizes astrocytomas to programmed cell death, and HTLV-1-infection induces the expression of IL-1β, IL-1α, IL-6, TNF-α, TNF-β [26]. A rat model for HTLV-1 infection demonstrated a role for apoptosis in the destruction of oligodendricytes and Schwann cells associated with the down-regulation of Bcl-2 and the up-regulation of Bax and p53 [27, 28]. Future work is needed to fully elucidate the roles of programmed cell death and the induction of a pro-inflammatory response in this chronic inflammatory disease.
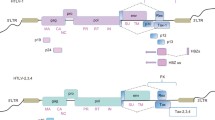
HTLV-1 exhibits several unique properties not seen in other animal onco-retroviruses. The end of the proviral genome contains several open reading frames encoding for the regulatory proteins p12, p30, p13 and HBZ (Fig. 1), which are involved in virus infectivity, immune escape, and the establishment of a latent state [29]. The viral protein Rex binds an RNA element (RxRE) present in the 3′ region of the viral mRNA and stimulates the transport of unspliced or singly spliced viral RNA to the cytoplasm to express structural proteins. Perhaps the most studied viral protein is the viral transcriptional transactivator Tax, which is involved in cellular transformation and specifically interacts with CREB, coactivators CBP/p300, and PCAF to stimulate transcription from the viral long terminal repeat (LTR) [30–34]. Tax plays an important role in the initiation of cellular transformation and also stimulates cellular proliferation by inactivating several cell cycle checkpoints [35–37]. In addition, several studies have shown that Tax inhibits the nucleotide excision repair (NER) pathway, beta-polymerase and topoisomerase [37–39]. While these events may facilitate cellular transformation, it is likely that cells need to acquire a pre-tumoral genotype and tolerance to Tax expression before transformation takes place.

Proteins encoded by HTLV type I. Multiple differentially spliced mRNA molecules transcribed from the genome of HTLV-1 encode for a dozen known proteins, with transcription initiated via the long terminal repeat (LTR). Homologues of proteins such as Gag, Pol (polymerase), Pro (protease), and Env (envelope protein) are also found in other retroviruses such as HIV, and are responsible for virus replication and virion formation. The remaining non-structural proteins characterized to date, such as Tax, Rex, p13, p12, p30, p21Rex and HBZ, are unique proteins translated from the pX region of the viral genome, and their localization is shown at right. Proteins shaded in grey have been shown to play either a direct or indirect role in modulating the apoptotic cascade in HTLV-1-infected cells
Recent studies have shown that the apoptotic pathway can be reactivated in HTLV-1-transformed cells, indicating that the apoptotic machinery is likely intact. It is the focus of this review to examine the underlying mechanisms HTLV-1 uses to repress apoptosis, and to highlight the therapies being evaluated to reactivate and trigger the apoptotic pathway in HTLV-1-transformed cells and infected patients.
HTLV-1 Tax: regulation of NF-κB, Akt, and gene expression
Tax is a potent trans-activator of transcription, and induces the constitutive activation of the major cellular pro-survival pathways NF-κB and Akt. Twenty years ago, it was first documented that Tax could induce transcription from the interleukin-2 gene via NF-κB related factors, indicating that Tax could activate NF-κB-regulated genes [40, 41]. It has since been demonstrated that Tax activates NF-κB through several different mechanisms. Tax can directly interact with IKKγ, ultimately triggering the continual phosphorylation and ubiquitin-mediated degradation of IκB to allow NF-κB translocation to the nucleus (Fig. 2) [42–45]. The direct activation of IKK by Tax has recently been demonstrated using an in vitro assay [46], and this activation step requires the phosphorylation of IKK. Alternatively, Tax can form a complex with the p100 NF-κB precursor protein along with IKKα/IKKγ to facilitate the cleavage of p100 into the active p52 NF-κB subunit [47]. Thirdly, Tax can interact directly with NF-κB subunits to facilitate NF-κB transcriptional activation [48–50], and has also been shown to directly recruit transcriptional co-activators CBP/p300 to NF-κB complexes in the nucleus [32, 51, 52].

Apoptotic regulatory pathways interrupted by HTLV-1 proteins. The viral oncoprotein Tax inactivates the inhibitors of κB through the activation of IKK, resulting in IκB phosphorylation and degradation, and the release of NF-κB. NF-κB is free to translocate to the nucleus to induce the transcription of pro-survival genes. Tax also stimulates the constitutive activation of Akt, resulting in the activation of β-catenin and AP-1 transcriptional pathways, leading to the up-regulation of additional anti-apoptotic genes. The small viral protein p12 has been shown to interact with the IL-2 receptor (IL-2R), thus stimulating Jak-recruitment. This leads to the phosphorylation, dimerization, and nuclear translocation of STAT5 to facilitate the up-regulation of pro-survival gene products. The HTLV-1 protein p13 localizes to the inner mitochondrial membrane where it may play a role in mitochondrial morphology and regulation of the permeability transition (PT) pore
The nuclear translocation and activation of NF-κB can lead to the transcriptional up-regulation of a number of anti-apoptotic proteins (Fig. 2). One potent anti-apoptotic protein up-regulated by Tax-mediated NF-κB and CREB activation is Bcl-xL [53, 54], and T-cells from HTLV-1-infected patients correspondingly display up-regulated levels of Bcl-xL [55]. In support of the role that NF-κB plays in the inhibition of cell death in HTLV-1 infected cells, drugs which inhibit NF-κB are potent inducers of tumor cell death in vitro [56] (Discussed below, see Table 1). The induction of NF-κB activation by Tax also increases expression of the inhibitor of apoptosis (IAP) family (Fig. 1) [57, 58]. IAPs are capable of directly binding to caspases, and can induce caspase degradation. Indeed, siRNA directed against one IAP, HIAP, greatly sensitized cells to apoptosis, suggesting HIAP expression may be important for Tax-mediated survival [58]. The cell regulatory protein p21 is also transactivated by Tax, and contributes to an anti-apoptotic phenotype of Tax-immortalized cells via the transactivation of NF-κB/CREB leading to the activation of anti-apoptotic genes [59]. The T-cell co-stimulatory molecule 4-1BB (TNFRSF9/CD137/ILA), which is involved in cell proliferation and survival, is also up-regulated by Tax, likely through NF-κB [60].
Another cell signaling pathway modulated by Tax is Akt, a pro-survival serine/threonine kinase that is constitutively activated in most ATLL patients [61]. Akt is phosphorylated on Serine473 in most ATLL patients, and Tax promotes this by interacting with and activating the upstream phosphatidylinositol-3-kinase (PI3K) [62, 63]. Activated Akt induces the downstream activation of additional transcription factors such as AP-1 and β-catenin [64] (Fig. 2), leading to Bcl-xL expression, p53 repression, and cell survival. Indeed, under specific conditions treatment of HTLV-1-infected cells with LY294002, an inhibitor of the PI3K pathway, induces cell death [61, 65], supporting the role that Akt plays in Tax-mediated cell survival. As well, certain reports have suggested that there is a cross-talk between Akt and NF-κB [61].
In addition to the activation of the NF-κB and Akt pathways, Tax also alters the transcription factor AP-1 [66, 67], although the specific effects of Tax-mediated AP-1 activation remain to be characterized. Tax also modulates a number of apoptotic genes via unknown mechanisms. Tax induces the production of cellular FLICE-inhibitory proteins (c-FLIPs) [68], which can inhibit CD95-induced cell death. HTLV-1 infection also induces the expression of the telomerase gene hTERT to protect transformed cells from replicative senescence [69]. Interestingly, hTERT also has the ability to inhibit mitochondrial cell death induced by specific pro-apoptotic stimuli [70]. Whether the induction of hTERT expression by Tax also has pro-survival effects at the mitochondria has yet to be explored. Recent microarray data demonstrated the general down-regulation of anti-apoptotic genes in HTLV-1-transformed cells [71] and that the induction of Akt/PI3K and the inactivation and phosphorylation of the pro-apoptotic Bcl-2 family member Bad might be critical to the regulation of apoptosis.
While Tax constitutively activates Akt and NF-κB, Tax also negatively regulates the cell cycle checkpoint tumor suppressor p53, which normally triggers cell cycle arrest and apoptosis in response to DNA damage [72]. p53 is functionally inactivated by Tax and is mutated in approximately 30% of all ATLL patients [73–75], thereby abrogating p53-mediated G1 cell cycle arrest and p53-mediated apoptosis in ATLL tumour cells [76]. Even in absence of genetic mutation, p53 appears to be inactivated in ATLL cells in vivo [77]. Tax-mediated inactivation of p53 is believed to occur through p53 phosphorylation on specific residues [74, 78]. As well, a recent study also suggests that the transcriptional repressor of p53, MdmX, is up-regulated in HTLV-1 infected cells in vitro and in vivo, and may play an important role in the inactivation of p53 in the absence of Tax expression [79]. The ability of Tax to repress the non-transcriptional functions of p53 is intriguing. Tax-mediated repression of p53 transactivation has a profound effect on G1 arrest and apoptosis induced by p53 overexpression [80]. In that study, the CREB/ATF, but not the NF-κB activation by Tax was essential for p53-inhibition [81]. The amount of protection from apoptosis obtained upon expression of Tax correlated with the decreased transcriptional activation of p53 observed in the various cell lines, indicating that the Tax-mediated protection from apoptosis may in part be related to the suppression of p53 transcriptional activity. Interestingly, p53 has also been shown to have a direct pro-apoptotic role at the mitochondria [82]. Whether this particular pro-apoptotic function is altered in ATLL or HTLV-1-infected cells is unknown.
In addition to p53, Tax has been shown to affect virtually every other cell cycle phase and checkpoint, including G1phase, G1/S checkpoint, S phase, G2/M checkpoint, and mitosis. Tax directly interacts with the cell cycle checkpoint kinase 2 (Chk2), and inhibits gamma-irradiation-induced apoptosis [17]. Additional effects of Tax on the cell cycle have been recently reviewed [36, 37].
The fact that Tax constitutively activates both NF-κB and Akt, and that Tax simultaneously inactivates p53 should point to a broad anti-apoptotic activity of Tax. Experimental data, however, remain controversial, as numerous studies have reported that the overexpression of Tax induces apoptosis. Over-expression of Tax sensitized cells to DNA-damage-induced apoptosis in a p53-independent manner [83, 84], and induced cell death in Jurkat cells expressing CD95 (Fas) in a caspase-dependent manner [85]. This Tax-induced death can be blocked by Bcl-2 expression [86]. Tax was also observed to induce caspase-dependent cell death that correlated with the ability of Tax to regulate p300/CBP activity, but not NF-κB activity [87]. These observations are similar to those seen with other oncogenic factors such as Myc, Cyclin D and E1A, which also display both proliferative and pro-apoptotic effects. In contrast to most viral proteins which directly inhibit a particular checkpoint of the apoptotic cascade, Tax alters the expression of cellular genes and hijacks cell signaling pathways. Therefore, cells of different origin expressing different proteins may respond in different ways to Tax expression adding confusion to the field. Additional factors that influence cellular fate are the levels and duration of Tax expression. Tax transgenic mice develop numerous tumors and cells isolated from those tumors are highly refractory to various apoptotic stimuli [56]. It is possible that Tax directly protects these tumor cells by inducing NF-κB or Akt activation or other pathways. On the other hand, it is also possible that Tax does exert an initial pro-apoptotic stimulus, and that tumor cells are derived from cells that have subsequently acquired resistance to Tax-induced pro-apoptotic signals. In support of such model, thymus atrophy has been reported in some transgenic models, and was also associated with massive amounts of apoptosis [88].
Although Tax appears to be required for cell transformation and the inhibition of apoptosis, ATLL tumor cells do not express detectable levels of Tax [89–91]. Surprisingly, ATLL tumor cells that lack Tax still retain the characteristics of Tax-expressing cells, and multiple signaling pathways such as NF-κB are constitutively active in ATLL cells. These observations suggest that following cell transformation, cellular signaling molecules remain permanently activated in the absence of Tax. This correlates with the requirement of Tax for the initial transformation event, but not for the maintenance of the transformed state.
ORFII: the mitochondrial p13 and the regulatory protein p30
Many viruses encode proteins that localize to the mitochondria to modulate this important apoptotic checkpoint, and HTLV-1 appears to be no exception. The small HTLV protein p13 is a small, 87 amino acid non-structural protein encoded by the XII open reading frame. p13 targets to mitochondria via an N-terminal mitochondrial targeting motif between amino acids 19 and 31 that allows p13 to insert into the inner mitochondrial membrane (Fig. 2) [92]. While most integral inner membrane proteins of the mitochondria possess classic signal sequences which are cleaved during protein import, p13 does not appear to be cleaved, leaving the mechanism of import unknown. The targeting motif is rich in arginine residues and is predicted to resemble an amphipathic alpha helix, similar to other mitochondrial proteins produced by RNA viruses. One such alpha-helical protein is the viroporin Vpr from HIV [93]. The amphipathic alpha-helical nature of Vpr allows it to form cation-selective channels in the mitochondria membrane. This results in mitochondrial depolarization [93], which is dependent on the mitochondrial permeability transition pore proteins ANT and VDAC that interact with Vpr [93]. Other viroporins include HBX from hepatitis B virus and PB1-F2 from Influenza A virus, which also localize to the mitochondria via short transmembrane domains and induce mitochondrial alterations leading to apoptosis [94–98]. Although Vpr and HBX induce cytochrome c release, there is no evidence to suggest that p13 similarly induces cytochrome c release. p13, however, does appear to sensitize cells to pro-apoptotic stimuli, as p13 expression has a dose-dependent effect on amplifying apoptosis induced by either anti-Fas or ceramide [99]. p13 directly interacts with farnesyl pyrophosphate synthetase, which catalyzes the generation of substrates involved in the Ras pathway [100]. Inclusion of a farnesyl transferase inhibitor that blocks Ras prenylation also blocks FasL- and ceramide-induced apoptosis in p13-expressing T-cells [99]. Exactly how p13 modulates apoptosis at the mitochondria is unknown, although biochemical studies showed that p13 expression induced the loss of the mitochondrial membrane potential and caused a decrease in the calcium retention capacity of mitochondria [101]. These events appear to be independent of the permeability transition (PT) pore, as the PT pore inhibitor cyclosporine A has no effect on p13-mediated PT [101]. This is in contrast with other viral mitochondrial pro-apoptotic proteins such as Vpr, which interact with components of the PT pore to directly induce PT [93]. It has been demonstrated that accumulation of p13 at the mitochondria results in the rounding and fragmentation of the mitochondrial network, and is associated with mitochondrial swelling and cristae fragmentation [101]. Substitution of glutamine for each of the four arginine residues present in the N-terminal alpha-helix has no effect on p13 localization, but prevents p13-dependent mitochondrial rearrangement and fragmentation [101]. This may be important since recent work has implicated the fission and fusion of the mitochondrial network in the regulation of apoptotic cell death [102–105]. How p13 controls mitochondrial morphology remains to be investigated. Future work using p13 mutants which localize to but do not induce mitochondrial rearrangements will help elucidate the mechanism used by p13 to modulate mitochondrial morphology and establish whether these morphological changes are required for the regulation of apoptosis or virus virulence.
Another viral protein synthesized from ORFII is p30, which is a post-transcriptional regulator of translation. p30 expression inhibits the translocation of Tax/Rex mRNA from the nucleus to the cytoplasm, thereby inhibiting Tax and Rex protein production [106]. While it remains to be seen, the expression of p30 may alter the ability of Tax and HTLV-1 to modulate programmed cell death. In cases where high Tax expression is detrimental to the cell, it is possible that the inhibition of Tax synthesis by p30 decreases the likelihood of apoptosis induction, thereby facilitating virus latency. Alternatively, since p30 selectively blocks mRNA nuclear export, p30 expression might somehow inhibit the pro-survival mechanisms that Tax uses, thereby sensitizing the cell to apoptosis. Microarray analysis examining the effect of p30 on cellular gene expression demonstrated that the expression of a number of apoptosis-related genes was altered, including genes encoding Mcl-1, A1, Bik, and caspases 2 and 4 [107]. Whether the regulation of any of these apoptotic genes is involved in the modulation of apoptosis by HTLV-1 remains to be investigated.
ORFI: p12I and IL-2R signaling survival pathway
A hallmark of HTLV-1 transformed cells is the constitutive activation of the Jak/STAT (Janus activating kinase/signal transducer and activator of transcription) pathway [108, 109], which rids infected lymphocytes of their dependence on IL-2 for proliferation and activation. Jak/STATs are involved in a number of cell processes, from cytokine signaling to the interferon response. Although various members of the Jak/STAT family have the potential to elicit both pro- and anti-apoptotic effects, one STAT, STAT5, specifically has anti-apoptotic effects [110, 111]. This includes the up-regulation of anti-apoptotic Bcl-2 family members such as Bcl-xL and Bcl-2, as well as the down-regulation of caspases 3 and 9 [110].
The HTLV-1 non-structural protein p12 from open reading frame I is critical for establishing viral infection in vivo [112, 113]. p12I enhances STAT5 activation by binding the β and γc chains of the IL-2 receptor, resulting in Jak1/Jak3 activation, STAT5a/b phosphorylation and nuclear translocation of the STAT5 heterodimer (Fig. 2). p12I increases STAT5 phosphorylation and STAT5 DNA binding in the absence of IL-2 [114]. The STAT5 activation induced by p12I appears to up-regulate X-linked IAP (XIAP), as the nucleoside analogue Roscovotine inhibits STAT5 and results in a decrease in XIAP expression in HTLV-1-infected cells [115].
HBZ: new player on the scene?
Recent research has characterized a novel protein transcribed from the negative strand of the HTLV-1 genome, HTLV-1 basic leucine-zipper factor, or HBZ [116]. This protein interacts with transcription factors CREB and those of the Jun family, and impairs the DNA binding ability of c-Jun [117–119]. As a result, HBZ has the ability to repress transcription of factors such as AP-1, Tax, and NF-κB. Although HBZ appears to play a repressive role in expression of certain cellular factors and viral genes, whether HBZ also affects the ability of Tax and other viral proteins to modulate the apoptotic cascade remains to be investigated.
Treatment of HTLV-1: drug-induced apoptosis
To date, a successful therapy for HTLV-1 has remained elusive in that many broad-range cancer therapies are ineffective. A wide range of combinatorial anti-cancer therapies have been used in clinical trials with limited degrees of success [120]. Recently, a number of new compounds and therapies have been shown to specifically induce apoptosis in HTLV-1 and ATLL cells (see Table 1), and many of these drugs target the aforementioned changes in gene expression and protein function that are essential for ATLL cell survival (Fig. 3).

Cellular pathways targeted by drugs which induce apoptosis in HTLV-1-infected cells. Drugs used to induce apoptosis in HTLV-1 and ATLL cells in vitro have targeted various aspects of the NF-κB pathway, such as inhibition of the proteasome, inhibition of the IKK complex, and inhibition of nuclear translocation of NF-κB. Other drugs have been used to target the cell cycle by stabilizing p53 or by inducing cell cycle arrest. Inhibitors of gene transcription have targeted STAT5 and the down-regulation of various anti-apoptotic proteins. More recently, a number of monoclonal antibody therapies have targeted cell surface proteins up-regulated in HTLV-1-infected cells, such as the IL-2 receptor, transferrin receptor, and CD52
Considering the importance of NF-κB in ATLL cell survival, one group of drugs being examined targets the NF-κB pathway. Bay 11-7082 and ACHP, inhibitors of IκB phosphorylation, and the proteasome inhibitor bortezomib/PS-341 inhibit both HTLV-1 and Tax-mediated NF-κB activation and induce apoptosis in infected cells [121–126]. Notably, cells treated with bortezomib/PS-341 in the presence of the caspase-inhibitor zVAD-fmk appear to undergo necrosis instead of apoptosis, indicating that the mechanism of death is still unclear [123]. The in vivo efficacy of bortezomib, however, remains to be seen, as one particular clinical trial demonstrated that one ATLL patient did not respond to bortezomib treatment [127]. The purine analogue Fludarabine also inhibits NF-κB activation resulting in the induction of apoptosis in HTLV-1-infected cells [128]. An HIV protease inhibitor, ritonavir, induces apoptosis in HTLV-1-leukemic cells by inhibiting NF-κB activity [129]. While ritonavir has not yet been tested for ATLL, it has shown efficacy in the treatment of HIV-related AIDS [130]. Altogether, the inhibition of NF-κB signaling appears to be a very promising target for new and developing HTLV-1-therapies. The role of NF-κB regulation in ATLL is not unique, as multiple lymphomas, such as Hodgkins disease, MALT lymphomas, and Kaposi’s sarcoma are associated with the deregulation of NF-κB activity [131].
Arsenic trioxide, alone or in combination with other pro-apoptotic stimuli, has also been examined as a possible treatment and induces apoptosis in HTLV-1-infected cells lines [132–134]. While arsenic induces the generation of hydrogen peroxide leading to cytochrome c release and caspase activation [135], recent work has also suggested that arsenic trioxide causes cell cycle arrest, NF-κB repression, and the down-regulation of Tax [132–134, 136–138]. Clinical use of arsenic, however, is problematic as there are differences in sensitivity to As2O3, and arsenic itself is toxic at high doses. Inclusion of polyunsaturated fatty acids such as docosahexaenoic acid (DHA), which increases ROS production and lipid peroxidation, and Emodin significantly increases necrotic cell death in HTLV-1-infected cells following treatment with As2O3 [139]. The use of combinatorial therapies with arsenic may allow for lower doses of As2O3 to be used.
Other cell signaling pathways that are deregulated in HTLV-1-infected cells have also been areas for drug development. The purine analogue roscovitine inhibits STAT5 activation and XIAP expression to induce apoptosis in MT-2 HTLV-1-infected cells [115]. Curcumin, a natural pigment of the spice turmeric, has been used extensively as an anticancer drug, and treatment of HTLV-1-infected cells with curcumin induces apoptosis by targeting the Akt-survival pathway or the Jak/STAT pathway [140–143]. Results suggesting that a specific Jak-inhibitor, AG-490, induces cell cycle arrest, however, are controversial [141, 144]. The geldanamycin derivative 17-AAG inhibits the activity of heat shock protein 90 (Hsp90), and is able to induce apoptosis in primary ATLL cells [145]. Although there is no clinical data for the use of 17-AAG in ATLL patients, 17-AAG has been successfully tested in a phase I clinical trial for various other malignancies [146].
ATLL cells are often characterized by the overexpression of specific cell surface markers, and a number of monoclonal antibodies have correspondingly been developed with the intention of inducing cell death. One early antibody therapy attempted was the use of anti-Fas [147–149]. Despite early success, however, the efficacy of anti-Fas in providing long-term remission was inadequate for clinical use. Other monoclonal antibody therapies directed at the overexpressed IL-2 receptor (CD25) have shown a greater degree of promise [150–152]. Early clinical trials with the antibody anti-Tac, which is directed against the IL-2α receptor, demonstrated limited success, although recent developments using a Yttrium-90-radiolabeled antibody has exhibited an increased activity against ATLL cells [150, 151, 153–155]. Anti-IL-2Rα antibody, in conjunction with bortezomib/PS-341 treatment was able to elicit the complete remission in ATLL-tumour-bearing mice [156], again demonstrating the efficacy of a combinatorial therapy. A major positive for anti-Tac therapy is the low level of side-effects, which is in stark contrast to standard chemotherapy reagents. More recently, other monoclonal antibodies have been directed at CD52 and the transferrin receptor, both of which are also overexpressed in HTLV-transformed cells [157–159].
Another class of pro-apoptotic drugs being investigated to treat ATLL targets the cell cycle. Retinoic acids induce apoptosis in HTLV-1-infected cells and ex vivo ATL cells [160–162], primarily by inducing cell cycle arrest. One retinoic acid, N-(4-hydroxyphenyl) retinamide, induced the dramatic death of malignant ATL, and was associated with elevated ceramide levels leading to cell cycle arrest and Bax activation [163]. Although there are specific effects on gene expression, these retinoids ultimately induce cell death through the mitochondrial pathway which is regulated by Bcl-2 [164]. A number of other retinoids have also been documented to induce apoptosis in HTLV-1-infected cells [165–169]. Perhaps the most-studied anti-retroviral drug is zidovudine (AZT), which is used extensively to treat HIV-1-infected individuals. Despite early reports suggesting that zidovudine provided some level of anti-cancer effect in ATL patients [170, 171], ATL cells do not appear to exhibit a high degree of apoptosis in response to zidovudine, even in combination with IFNα [172], and the mechanism of inhibition is likely through telomere attrition and reactivation of a p53-dependent senescent pathway [79, 173, 174].
Considering the extensive work performed in pursuit of new potential therapies, it is of note that certain members of the multi-drug resistance (MDR) protein family are up-regulated in HTLV-1-infected cells and ATLL patients [175–177]. Adaptations such as these may dictate the relative sensitivity to various drug therapies for ATLL patients, and should be noted when promising new emerging therapies are investigated.
Concluding remarks
Like many viruses, HTLV-1 modulates the apoptotic pathway using multiple tactics, ranging from Tax-mediated modulation of gene expression and the cell cycle, to STAT activation by p12, to the regulation of the mitochondria by p13. Future work will hopefully further expose the specific mechanisms used by HTLV-1 to control cell death, and these investigations will aid in our understanding of the pathology of HTLV-1-infection and ensuing ATLL. Therapeutic strategies aimed at inducing virus-infected cell death must consider the fact that not all deaths are equal. Necrotic cell death results in an inflammatory response, while apoptosis, in contrast, is a tightly controlled process that does not lead to inflammation. The ability of HTLV-1-infected ATLL cells to resist apoptosis likely greatly contributes to the development of ATLL. In contrast, the pathogenesis of TSP/HAM is associated with high inflammation and hyper-immune responses [12]. Taking these findings into considerations, whether it is beneficial to induce either apoptosis in TSP/HAM and necrotic cell death in ATLL patients has not been addressed. The development of new therapies to treat HTLV-1-infected patients diagnosed with ATLL or TSP/HAM may hinge upon the ability of new drugs or combinatorial therapies to specifically induce death in HTLV-1-infected T cells in vivo.
References
Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26:239–257
Thompson CB (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267:1456–1462
Hardwick JM, Bellows DS (2003) Viral versus cellular BCL-2 proteins. Cell Death Differ 10(Suppl 1):S68–S76
Benedict CA, Norris PS, Ware CF (2002) To kill or be killed: viral evasion of apoptosis. Nat Immunol 3:1013–1018
Boya P, Pauleau AL, Poncet D, Gonzalez-Polo RA, Zamzami N, Kroemer G (2004) Viral proteins targeting mitochondria: controlling cell death. Biochim Biophys Acta 1659:178–189
Roulston A, Marcellus RC, Branton PE (1999) Viruses and apoptosis. Annu Rev Microbiol 53:577–628
Taylor JM, Barry M (2006) Near death experiences: poxvirus regulation of apoptotic death. Virology 344:139–150
Danthi P, Kobayashi T, Holm GH, Hansberger MW, Abel TW, Dermody TS (2008) Reovirus apoptosis and virulence are regulated by host cell membrane penetration efficiency. J Virol 82:161–172
Eckardt-Michel J, Lorek M, Baxmann D, Grunwald T, Keil GM, Zimmer G (2008) The fusion protein of respiratory syncytial virus triggers p53-dependent apoptosis. J Virol 82:3236–3249
Franchini G (1995) Molecular mechanisms of human T-cell leukemia/lymphotropic virus type I infection. Blood 86:3619–3639
Nicot C (2005) Current views in HTLV-I-associated adult T-cell leukemia/lymphoma. Am J Hematol 78:232–239
Kiwaki T, Umehara F, Arimura Y, Izumo S, Arimura K, Itoh K et al (2003) The clinical and pathological features of peripheral neuropathy accompanied with HTLV-I associated myelopathy. J Neurol Sci 206:17–21
Nakagawa M, Izumo S, Ijichi S, Kubota H, Arimura K, Kawabata M et al (1995) HTLV-I-associated myelopathy: analysis of 213 patients based on clinical features and laboratory findings. J Neurovirol 1:50–61
Yang YC, Hsu TY, Lin RH, Su IJ, Chen JY, Yang CS (2002) Resistance to tumor necrosis factor-alpha-induced apoptosis in human T-lymphotropic virus type I-infected T cell lines. AIDS Res Hum Retroviruses 18:207–212
Copeland KF, Haaksma AG, Goudsmit J, Krammer PH, Heeney JL (1994) Inhibition of apoptosis in T cells expressing human T cell leukemia virus type I Tax. AIDS Res Hum Retroviruses 10:1259–1268
Kishi S, Saijyo S, Arai M, Karasawa S, Ueda S, Kannagi M et al (1997) Resistance to fas-mediated apoptosis of peripheral T cells in human T lymphocyte virus type I (HTLV-I) transgenic mice with autoimmune arthropathy. J Exp Med 186:57–64
Park HU, Jeong SJ, Jeong JH, Chung JH, Brady JN (2006) Human T-cell leukemia virus type 1 Tax attenuates gamma-irradiation-induced apoptosis through physical interaction with Chk2. Oncogene 25:438–447
Hasegawa H, Yamada Y, Harasawa H, Tsuji T, Murata K, Sugahara K et al (2005) Sensitivity of adult T-cell leukaemia lymphoma cells to tumour necrosis factor-related apoptosis-inducing ligand. Br J Haematol 128:253–265
Tsuda H, Huang RW, Takatsuki K (1993) Interleukin-2 prevents programmed cell death in adult T-cell leukemia cells. Jpn J Cancer Res 84:431–437
Tamiya S, Etoh K, Suzushima H, Takatsuki K, Matsuoka M (1998) Mutation of CD95 (Fas/Apo-1) gene in adult T-cell leukemia cells. Blood 91:3935–3942
Maeda T, Yamada Y, Moriuchi R, Sugahara K, Tsuruda K, Joh T et al (1999) Fas gene mutation in the progression of adult T cell leukemia. J Exp Med 189:1063–1071
Arai M, Kannagi M, Matsuoka M, Sato T, Yamamoto N, Fujii M (1998) Expression of FAP-1 (Fas-associated phosphatase) and resistance to Fas-mediated apoptosis in T cell lines derived from human T cell leukemia virus type 1-associated myelopathy/tropical spastic paraparesis patients. AIDS Res Hum Retroviruses 14:261–267
Hamasaki S, Nakamura T, Furuya T, Kawakami A, Ichinose K, Nakashima T et al (2001) Resistance of CD4-positive T lymphocytes to etoposide-induced apoptosis mediated by upregulation of Bcl-xL expression in patients with HTLV-I-associated myelopathy. J Neuroimmunol 117:143–148
Kawahigashi N, Furukawa Y, Saito M, Usuku K, Osame M (1998) Predominant expression of Fas ligand mRNA in CD8+ T lymphocytes in patients with HTLV-1 associated myelopathy. J Neuroimmunol 90:199–206
Chen X, Zachar V, Zdravkovic M, Guo M, Ebbesen P, Liu X (1997) Role of the Fas/Fas ligand pathway in apoptotic cell death induced by the human T cell lymphotropic virus type I Tax transactivator. J Gen Virol 78( Pt 12):3277–3285
Banerjee P, Rochford R, Antel J, Canute G, Wrzesinski S, Sieburg M et al (2007) Proinflammatory cytokine gene induction by human T-cell leukemia virus type 1 (HTLV-1) and HTLV-2 Tax in primary human glial cells. J Virol 81:1690–1700
Tomaru U, Ikeda H, Jiang X, Ohya O, Yoshiki T (2003) Provirus expansion and deregulation of apoptosis-related genes in the spinal cord of a rat model for human T-lymphocyte virus type I-associated myeloneuropathy. J Neurovirol 9:530–538
Ohya O, Tomaru U, Yamashita I, Kasai T, Morita K, Ikeda H et al (1997) HTLV-I induced myeloneuropathy in WKAH rats: apoptosis and local activation of the HTLV-I pX and TNF-alpha genes implicated in the pathogenesis. Leukemia 11(Suppl 3):255–257
Nicot C, Harrod RL, Ciminale V, Franchini G (2005) Human T-cell leukemia/lymphoma virus type 1 nonstructural genes and their functions. Oncogene 24:6026–6034
Zhao LJ, Giam CZ (1992) Human T-cell lymphotropic virus type I (HTLV-I) transcriptional activator, Tax, enhances CREB binding to HTLV-I 21-base-pair repeats by protein-protein interaction. Proc Natl Acad Sci USA 89:7070–7074
Suzuki T, Fujisawa JI, Toita M, Yoshida M (1993) The trans-activator tax of human T-cell leukemia virus type 1 (HTLV-1) interacts with cAMP-responsive element (CRE) binding and CRE modulator proteins that bind to the 21-base-pair enhancer of HTLV-1. Proc Natl Acad Sci USA 90:610–614
Bex F, Yin MJ, Burny A, Gaynor RB (1998) Differential transcriptional activation by human T-cell leukemia virus type 1 Tax mutants is mediated by distinct interactions with CREB binding protein and p300. Mol Cell Biol 18:2392–2405
Jiang H, Lu H, Schiltz RL, Pise-Masison CA, Ogryzko VV, Nakatani Y et al (1999) PCAF interacts with tax and stimulates tax transactivation in a histone acetyltransferase-independent manner. Mol Cell Biol 19:8136–8145
Franklin AA, Kubik MF, Uittenbogaard MN, Brauweiler A, Utaisincharoen P, Matthews MA et al (1993) Transactivation by the human T-cell leukemia virus Tax protein is mediated through enhanced binding of activating transcription factor-2 (ATF-2) ATF-2 response and cAMP element-binding protein (CREB). J Biol Chem 268:21225–21231
Hall WW, Fujii M (2005) Deregulation of cell-signaling pathways in HTLV-1 infection. Oncogene 24:5965–5975
Grassmann R, Aboud M, Jeang KT (2005) Molecular mechanisms of cellular transformation by HTLV-1 Tax. Oncogene 24:5976–5985
Marriott SJ, Semmes OJ (2005) Impact of HTLV-I Tax on cell cycle progression and the cellular DNA damage repair response. Oncogene 24:5986–5995
Suzuki T, Uchida-Toita M, Andoh T, Yoshida M (2000) HTLV-1 tax oncoprotein binds to DNA topoisomerase I and inhibits its catalytic activity. Virology 270:291–298
Jeang KT, Widen SG, Semmes OJ, Wilson SH (1990) HTLV-I trans-activator protein, tax, is a trans-repressor of the human beta-polymerase gene. Science 247:1082–1084
Ballard DW, Bohnlein E, Lowenthal JW, Wano Y, Franza BR, Greene WC (1988) HTLV-I tax induces cellular proteins that activate the kappa B element in the IL-2 receptor alpha gene. Science 241:1652–1655
Leung K, Nabel GJ (1988) HTLV-1 transactivator induces interleukin-2 receptor expression through an NF-kappa B-like factor. Nature 333:776–778
Iha H, Kibler KV, Yedavalli VR, Peloponese JM, Haller K, Miyazato A et al (2003) Segregation of NF-kappaB activation through NEMO/IKKgamma by Tax and TNFalpha: implications for stimulus-specific interruption of oncogenic signaling. Oncogene 22:8912–8923
Lacoste J, Petropoulos L, Pepin N, Hiscott J (1995) Constitutive phosphorylation and turnover of I kappa B alpha in human T-cell leukemia virus type I-infected and Tax-expressing T cells. J Virol 69:564–569
Sun SC, Elwood J, Beraud C, Greene WC (1994) Human T-cell leukemia virus type I Tax activation of NF-kappa B/Rel involves phosphorylation and degradation of I kappa B alpha and RelA (p65)-mediated induction of the c-rel gene. Mol Cell Biol 14:7377–7384
Maggirwar SB, Harhaj E, Sun SC (1995) Activation of NF-kappa B/Rel by Tax involves degradation of I kappa B alpha and is blocked by a proteasome inhibitor. Oncogene 11:993–998
Mukherjee S, Negi VS, Keitany G, Tanaka Y, Orth K (2008) In vitro activation of the IKK complex by HTLV-1 Tax. J Biol Chem (epub ahead of print), January 26, 2008
Xiao G, Cvijic ME, Fong A, Harhaj EW, Uhlik MT, Waterfield M et al (2001) Retroviral oncoprotein Tax induces processing of NF-kappaB2/p100 in T cells: evidence for the involvement of IKKalpha. EMBO J 20:6805–6815
Beraud C, Sun SC, Ganchi P, Ballard DW, Greene WC (1994) Human T-cell leukemia virus type I Tax associates with and is negatively regulated by the NF-kappa B2 p100 gene product: implications for viral latency. Mol Cell Biol 14:1374–1382
Suzuki T, Hirai H, Fujisawa J, Fujita T, Yoshida M (1993) A trans-activator Tax of human T-cell leukemia virus type 1 binds to NF-kappa B p50 and serum response factor (SRF) and associates with enhancer DNAs of the NF-kappa B site and CArG box. Oncogene 8:2391–2397
Hirai H, Fujisawa J, Suzuki T, Ueda K, Muramatsu M, Tsuboi A et al (1992) Transcriptional activator Tax of HTLV-1 binds to the NF-kappa B precursor p105. Oncogene 7:1737–1742
Suzuki T, Hirai H, Yoshida M (1994) Tax protein of HTLV-1 interacts with the Rel homology domain of NF-kappa B p65 and c-Rel proteins bound to the NF-kappa B binding site and activates transcription. Oncogene 9:3099–3105
Bex F, McDowall A, Burny A, Gaynor R (1997) The human T-cell leukemia virus type 1 transactivator protein Tax colocalizes in unique nuclear structures with NF-kappaB proteins. J Virol 71:3484–3497
Mori N, Fujii M, Cheng G, Ikeda S, Yamasaki Y, Yamada Y et al (2001) Human T-cell leukemia virus type I tax protein induces the expression of anti-apoptotic gene Bcl-xL in human T-cells through nuclear factor-kappaB and c-AMP responsive element binding protein pathways. Virus Genes 22:279–287
Tsukahara T, Kannagi M, Ohashi T, Kato H, Arai M, Nunez G et al (1999) Induction of Bcl-x(L) expression by human T-cell leukemia virus type 1 Tax through NF-kappaB in apoptosis-resistant T-cell transfectants with Tax. J Virol 73:7981–7987
Nicot C, Mahieux R, Takemoto S, Franchini G (2000) Bcl-X(L) is up-regulated by HTLV-I and HTLV-II in vitro and in ex vivo ATLL samples. Blood 96:275–281
Portis T, Harding JC, Ratner L (2001) The contribution of NF-kappa B activity to spontaneous proliferation and resistance to apoptosis in human T-cell leukemia virus type 1 Tax-induced tumors. Blood 98:1200–1208
Kawakami A, Nakashima T, Sakai H, Urayama S, Yamasaki S, Hida A et al (1999) Inhibition of caspase cascade by HTLV-I tax through induction of NF-kappaB nuclear translocation. Blood 94:3847–3854
Waldele K, Silbermann K, Schneider G, Ruckes T, Cullen BR, Grassmann R (2006) Requirement of the human T-cell leukemia virus (HTLV-1) tax-stimulated HIAP-1 gene for the survival of transformed lymphocytes. Blood 107:4491–4499
Kawata S, Ariumi Y, Shimotohno K (2003) p21(Waf1/Cip1/Sdi1) prevents apoptosis as well as stimulates growth in cells transformed or immortalized by human T-cell leukemia virus type 1-encoded tax. J Virol 77:7291–7299
Pichler K, Kattan T, Gentzsch J, Kress AK, Taylor GP, Bangham CR et al (2008) Strong induction of 4-1BB, a growth and survival promoting costimulatory receptor, in HTLV-1-infected cultured and patients’ T-cells by the viral Tax oncoprotein. Blood (epub ahead of print), February 14, 2008
Jeong SJ, Pise-Masison CA, Radonovich MF, Park HU, Brady JN (2005) Activated AKT regulates NF-kappaB activation, p53 inhibition and cell survival in HTLV-1-transformed cells. Oncogene 24:6719–6728
Liu Y, Wang Y, Yamakuchi M, Masuda S, Tokioka T, Yamaoka S et al (2001) Phosphoinositide-3 kinase-PKB/Akt pathway activation is involved in fibroblast Rat-1 transformation by human T-cell leukemia virus type I tax. Oncogene 20:2514–2526
Peloponese JM Jr, Jeang KT (2006) Role for Akt/protein kinase B and activator protein-1 in cellular proliferation induced by the human T-cell leukemia virus type 1 tax oncoprotein. J Biol Chem 281:8927–8938
Tomita M, Kikuchi A, Akiyama T, Tanaka Y, Mori N (2006) Human T-cell leukemia virus type 1 tax dysregulates beta-catenin signaling. J Virol 80:10497–10505
Ikezoe T, Nishioka C, Bandobashi K, Yang Y, Kuwayama Y, Adachi Y et al (2007) Longitudinal inhibition of PI3K/Akt/mTOR signaling by LY294002 and rapamycin induces growth arrest of adult T-cell leukemia cells. Leuk Res 31:673–682
Mori N, Fujii M, Iwai K, Ikeda S, Yamasaki Y, Hata T et al (2000) Constitutive activation of transcription factor AP-1 in primary adult T-cell leukemia cells. Blood 95:3915–3921
Fujii M, Niki T, Mori T, Matsuda T, Matsui M, Nomura N et al (1991) HTLV-1 Tax induces expression of various immediate early serum responsive genes. Oncogene 6:1023–1029
Krueger A, Fas SC, Giaisi M, Bleumink M, Merling A, Stumpf C et al (2006) HTLV-1 Tax protects against CD95-mediated apoptosis by induction of the cellular FLICE-inhibitory protein (c-FLIP). Blood 107:3933–3939
Sinha-Datta U, Horikawa I, Michishita E, Datta A, Sigler-Nicot JC, Brown M et al (2004) Transcriptional activation of hTERT through the NF-kappaB pathway in HTLV-I-transformed cells. Blood 104:2523–2531
Massard C, Zermati Y, Pauleau AL, Larochette N, Metivier D, Sabatier L et al (2006) hTERT: a novel endogenous inhibitor of the mitochondrial cell death pathway. Oncogene 25:4505–4514
Akl H, Badran BM, Zein NE, Bex F, Sotiriou C, Willard-Gallo KE et al (2007) HTLV-I infection of WE17/10 CD4+ cell line leads to progressive alteration of Ca2+ influx that eventually results in loss of CD7 expression and activation of an antiapoptotic pathway involving AKT and BAD which paves the way for malignant transformation. Leukemia 21:788–796
Vousden KH, Lane DP (2007) p53 in health and disease. Nat Rev Mol Cell Biol 8:275–283
Mahieux R, Pise-Masison CA, Nicot C, Green P, Hall WW, Brady JN (2000) Inactivation of p53 by HTLV type 1 and HTLV type 2 Tax trans-activators. AIDS Res Hum Retroviruses 16:1677–1681
Pise-Masison CA, Mahieux R, Radonovich M, Jiang H, Duvall J, Guillerm C et al (2000) Insights into the molecular mechanism of p53 inhibition by HTLV type 1 Tax. AIDS Res Hum Retroviruses 16:1669–1675
Nagai H, Kinoshita T, Imamura J, Murakami Y, Hayashi K, Mukai K et al (1991) Genetic alteration of p53 in some patients with adult T-cell leukemia. Jpn J Cancer Res 82:1421–1427
Mesnard JM, Devaux C (1999) Multiple control levels of cell proliferation by human T-cell leukemia virus type 1 Tax protein. Virology 257:277–284
Takemoto S, Trovato R, Cereseto A, Nicot C, Kislyakova T, Casareto L et al (2000) p53 stabilization and functional impairment in the absence of genetic mutation or the alteration of the p14(ARF)-MDM2 loop in ex vivo and cultured adult T-cell leukemia/lymphoma cells. Blood 95:3939–3944
Pise-Masison CA, Brady JN (2005) Setting the stage for transformation: HTLV-1 Tax inhibition of p53 function. Front Biosci 10:919–930
Datta A, Nicot C (2008) Telomere attrition induces a DNA double-strand break damage signal that reactivates p53 transcription in HTLV-I leukemic cells. Oncogene 27:1135–1141
Haoudi A, Semmes OJ (2003) The HTLV-1 tax oncoprotein attenuates DNA damage induced G1 arrest and enhances apoptosis in p53 null cells. Virology 305:229–239
Mulloy JC, Kislyakova T, Cereseto A, Casareto L, LoMonico A, Fullen J et al (1998) Human T-cell lymphotropic/leukemia virus type 1 Tax abrogates p53-induced cell cycle arrest and apoptosis through its CREB/ATF functional domain. J Virol 72:8852–8860
Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M et al (2004) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303:1010–1014
Kao SY, Lemoine FJ, Mariott SJ (2000) HTLV-1 Tax protein sensitizes cells to apoptotic cell death induced by DNA damaging agents. Oncogene 19:2240–2248
Kao SY, Lemoine FJ, Marriott SJ (2001) p53-independent induction of apoptosis by the HTLV-I tax protein following UV irradiation. Virology 291:292–298
Chlichlia K, Busslinger M, Peter ME, Walczak H, Krammer PH, Schirrmacher V et al (1997) ICE-proteases mediate HTLV-I Tax-induced apoptotic T-cell death. Oncogene 14:2265–2272
Yamada T, Yamaoka S, Goto T, Nakai M, Tsujimoto Y, Hatanaka M (1994) The human T-cell leukemia virus type I Tax protein induces apoptosis which is blocked by the Bcl-2 protein. J Virol 68:3374–3379
Nicot C, Harrod R (2000) Distinct p300-responsive mechanisms promote caspase-dependent apoptosis by human T-cell lymphotropic virus type 1 Tax protein. Mol Cell Biol 20:8580–8589
Hall AP, Irvine J, Blyth K, Cameron ER, Onions DE, Campbell ME (1998) Tumours derived from HTLV-I tax transgenic mice are characterized by enhanced levels of apoptosis and oncogene expression. J Pathol 186:209–214
Franchini G, Wong-Staal F, Gallo RC (1984) Human T-cell leukemia virus (HTLV-I) transcripts in fresh and cultured cells of patients with adult T-cell leukemia. Proc Natl Acad Sci USA 81:6207–6211
Kinoshita T, Shimoyama M, Tobinai K, Ito M, Ito S, Ikeda S et al (1989) Detection of mRNA for the tax1/rex1 gene of human T-cell leukemia virus type I in fresh peripheral blood mononuclear cells of adult T-cell leukemia patients and viral carriers by using the polymerase chain reaction. Proc Natl Acad Sci USA 86:5620–5624
Tendler CL, Greenberg SJ, Blattner WA, Manns A, Murphy E, Fleisher T et al (1990) Transactivation of interleukin 2 and its receptor induces immune activation in human T-cell lymphotropic virus type I-associated myelopathy: pathogenic implications and a rationale for immunotherapy. Proc Natl Acad Sci USA 87:5218–5222
Ciminale V, Zotti L, D’Agostino DM, Ferro T, Casareto L, Franchini G et al (1999) Mitochondrial targeting of the p13II protein coded by the x-II ORF of human T-cell leukemia/lymphotropic virus type I (HTLV-I). Oncogene 18:4505–4514
Jacotot E, Ravagnan L, Loeffler M, Ferri KF, Vieira HL, Zamzami N et al (2000) The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J Exp Med 191:33–46
Su F, Schneider RJ (1997) Hepatitis B virus HBx protein sensitizes cells to apoptotic killing by tumor necrosis factor alpha. Proc Natl Acad Sci USA 94:8744–8749
Takada S, Shirakata Y, Kaneniwa N, Koike K (1999) Association of hepatitis B virus X protein with mitochondria causes mitochondrial aggregation at the nuclear periphery, leading to cell death. Oncogene 18:6965–6973
Shirakata Y, Koike K (2003) Hepatitis B virus X protein induces cell death by causing loss of mitochondrial membrane potential. J Biol Chem 278:22071–22078
Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I et al (2001) A novel influenza A virus mitochondrial protein that induces cell death. Nat Med 7:1306–1312
Gibbs JS, Malide D, Hornung F, Bennink JR, Yewdell JW (2003) The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J Virol 77:7214–7224
Hiraragi H, Michael B, Nair A, Silic-Benussi M, Ciminale V, Lairmore M (2005) Human T-lymphotropic virus type 1 mitochondrion-localizing protein p13II sensitizes Jurkat T cells to Ras-mediated apoptosis. J Virol 79:9449–9457
Lefebvre L, Vanderplasschen A, Ciminale V, Heremans H, Dangoisse O, Jauniaux JC et al (2002) Oncoviral bovine leukemia virus G4 and human T-cell leukemia virus type 1 p13(II) accessory proteins interact with farnesyl pyrophosphate synthetase. J Virol 76:1400–1414
D’Agostino DM, Silic-Benussi M, Hiraragi H, Lairmore MD, Ciminale V (2005) The human T-cell leukemia virus type 1 p13II protein: effects on mitochondrial function and cell growth. Cell Death Differ 12(Suppl 1):905–915
Cereghetti GM, Scorrano L (2006) The many shapes of mitochondrial death. Oncogene 25:4717–4724
Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ (2006) Role of Bax and Bak in mitochondrial morphogenesis. Nature 443:658–662
Scorrano L (2005) Proteins that fuse and fragment mitochondria in apoptosis: con-fissing a deadly con-fusion? J Bioenerg Biomembr 37:165–170
Sugioka R, Shimizu S, Tsujimoto Y (2004) Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem 279:52726–52734
Nicot C, Dundr M, Johnson JM, Fullen JR, Alonzo N, Fukumoto R et al (2004) HTLV-1-encoded p30II is a post-transcriptional negative regulator of viral replication. Nat Med 10:197–201
Michael B, Nair AM, Hiraragi H, Shen L, Feuer G, Boris-Lawrie K et al (2004) Human T lymphotropic virus type-1 p30II alters cellular gene expression to selectively enhance signaling pathways that activate T lymphocytes. Retrovirology 1:39
Migone TS, Lin JX, Cereseto A, Mulloy JC, O’Shea JJ, Franchini G et al (1995) Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I. Science 269:79–81
Takemoto S, Mulloy JC, Cereseto A, Migone TS, Patel BK, Matsuoka M et al (1997) Proliferation of adult T cell leukemia/lymphoma cells is associated with the constitutive activation of JAK/STAT proteins. Proc Natl Acad Sci USA 94:13897–13902
Debierre-Grockiego F (2004) Anti-apoptotic role of STAT5 in haematopoietic cells and in the pathogenesis of malignancies. Apoptosis 9:717–728
Collins ND, D’Souza C, Albrecht B, Robek MD, Ratner L, Ding W et al (1999) Proliferation response to interleukin-2 and Jak/Stat activation of T cells immortalized by human T-cell lymphotropic virus type 1 is independent of open reading frame I expression. J Virol 73:9642–9649
Albrecht B, Collins ND, Burniston MT, Nisbet JW, Ratner L, Green PL et al (2000) Human T-lymphotropic virus type 1 open reading frame I p12(I) is required for efficient viral infectivity in primary lymphocytes. J Virol 74:9828–9835
Collins ND, Newbound GC, Albrecht B, Beard JL, Ratner L, Lairmore MD (1998) Selective ablation of human T-cell lymphotropic virus type 1 p12I reduces viral infectivity in vivo. Blood 91:4701–4707
Nicot C, Mulloy JC, Ferrari MG, Johnson JM, Fu K, Fukumoto R et al (2001) HTLV-1 p12(I) protein enhances STAT5 activation and decreases the interleukin-2 requirement for proliferation of primary human peripheral blood mononuclear cells. Blood 98:823–829
Mohapatra S, Chu B, Wei S, Djeu J, Epling-Burnette PK, Loughran T et al (2003) Roscovitine inhibits STAT5 activity and induces apoptosis in the human leukemia virus type 1-transformed cell line MT-2. Cancer Res 63:8523–8530
Gaudray G, Gachon F, Basbous J, Biard-Piechaczyk M, Devaux C, Mesnard JM (2002) The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J Virol 76:12813–12822
Basbous J, Arpin C, Gaudray G, Piechaczyk M, Devaux C, Mesnard JM (2003) The HBZ factor of human T-cell leukemia virus type I dimerizes with transcription factors JunB and c-Jun and modulates their transcriptional activity. J Biol Chem 278:43620–43627
Matsumoto J, Ohshima T, Isono O, Shimotohno K (2005) HTLV-1 HBZ suppresses AP-1 activity by impairing both the DNA-binding ability and the stability of c-Jun protein. Oncogene 24:1001–1010
Lemasson I, Lewis MR, Polakowski N, Hivin P, Cavanagh MH, Thebault S et al (2007) Human T-cell leukemia virus type 1 (HTLV-1) bZIP protein interacts with the cellular transcription factor CREB to inhibit HTLV-1 transcription. J Virol 81:1543–1553
Taylor GP, Matsuoka M (2005) Natural history of adult T-cell leukemia/lymphoma and approaches to therapy. Oncogene 24:6047–6057
Mori N, Yamada Y, Ikeda S, Yamasaki Y, Tsukasaki K, Tanaka Y et al (2002) Bay 11-7082 inhibits transcription factor NF-kappaB and induces apoptosis of HTLV-I-infected T-cell lines and primary adult T-cell leukemia cells. Blood 100:1828–1834
Sanda T, Asamitsu K, Ogura H, Iida S, Utsunomiya A, Ueda R et al (2006) Induction of cell death in adult T-cell leukemia cells by a novel IkappaB kinase inhibitor. Leukemia 20:590–598
Satou Y, Nosaka K, Koya Y, Yasunaga JI, Toyokuni S, Matsuoka M (2004) Proteasome inhibitor, bortezomib, potently inhibits the growth of adult T-cell leukemia cells both in vivo and in vitro. Leukemia 18:1357–1363
Mitra-Kaushik S, Harding JC, Hess JL, Ratner L (2004) Effects of the proteasome inhibitor PS-341 on tumor growth in HTLV-1 Tax transgenic mice and Tax tumor transplants. Blood 104:802–809
Nasr R, El-Sabban ME, Karam JA, Dbaibo G, Kfoury Y, Arnulf B et al (2005) Efficacy and mechanism of action of the proteasome inhibitor PS-341 in T-cell lymphomas and HTLV-I associated adult T-cell leukemia/lymphoma. Oncogene 24:419–430
Palombella VJ, Conner EM, Fuseler JW, Destree A, Davis JM, Laroux FS et al (1998) Role of the proteasome and NF-kappaB in streptococcal cell wall-induced polyarthritis. Proc Natl Acad Sci USA 95:15671–15676
Strauss SJ, Maharaj L, Hoare S, Johnson PW, Radford JA, Vinnecombe S et al (2006) Bortezomib therapy in patients with relapsed or refractory lymphoma: potential correlation of in vitro sensitivity and tumor necrosis factor alpha response with clinical activity. J Clin Oncol 24:2105–2112
Nishioka C, Ikezoe T, Yang J, Koeffler HP, Taguchi H (2007) Fludarabine induces apoptosis of human T-cell leukemia virus type 1-infected T cells via inhibition of the nuclear factor-kappaB signal pathway. Leukemia 21:1044–1049
Dewan MZ, Uchihara JN, Terashima K, Honda M, Sata T, Ito M et al (2006) Efficient intervention of growth and infiltration of primary adult T-cell leukemia cells by an HIV protease inhibitor, ritonavir. Blood 107:716–724
De Clercq E (2004) Antiviral drugs in current clinical use. J Clin Virol 30:115–133
Jost PJ, Ruland J (2007) Aberrant NF-kappaB signaling in lymphoma: mechanisms, consequences, and therapeutic implications. Blood 109:2700–2707
Nasr R, Rosenwald A, El-Sabban ME, Arnulf B, Zalloua P, Lepelletier Y et al (2003) Arsenic/interferon specifically reverses 2 distinct gene networks critical for the survival of HTLV-1-infected leukemic cells. Blood 101:4576–4582
Mahieux R, Pise-Masison C, Gessain A, Brady JN, Olivier R, Perret E et al (2001) Arsenic trioxide induces apoptosis in human T-cell leukemia virus type 1- and type 2-infected cells by a caspase-3-dependent mechanism involving Bcl-2 cleavage. Blood 98:3762–3769
Mahieux R, Hermine O (2005) In vivo and in vitro treatment of HTLV-1 and HTLV-2 infected cells with arsenic trioxide and interferon-alpha. Leuk Lymphoma 46:347–355
Chen YC, Lin-Shiau SY, Lin JK (1998) Involvement of reactive oxygen species and caspase 3 activation in arsenite-induced apoptosis. J Cell Physiol 177:324–333
El-Sabban ME, Nasr R, Dbaibo G, Hermine O, Abboushi N, Quignon F et al (2000) Arsenic-interferon-alpha-triggered apoptosis in HTLV-I transformed cells is associated with tax down-regulation and reversal of NF-kappa B activation. Blood 96:2849–2855
Bazarbachi A, El-Sabban ME, Nasr R, Quignon F, Awaraji C, Kersual J et al (1999) Arsenic trioxide and interferon-alpha synergize to induce cell cycle arrest and apoptosis in human T-cell lymphotropic virus type I-transformed cells. Blood 93:278–283
Ishitsuka K, Hanada S, Uozumi K, Utsunomiya A, Arima T (2000) Arsenic trioxide and the growth of human T-cell leukemia virus type I infected T-cell lines. Leuk Lymphoma 37:649–655
Brown M, Bellon M, Nicot C (2007) Emodin and DHA potently increase arsenic trioxide interferon-alpha-induced cell death of HTLV-I-transformed cells by generation of reactive oxygen species and inhibition of Akt and AP-1. Blood 109:1653–1659
Rajasingh J, Raikwar HP, Muthian G, Johnson C, Bright JJ (2006) Curcumin induces growth-arrest and apoptosis in association with the inhibition of constitutively active JAK-STAT pathway in T cell leukemia. Biochem Biophys Res Commun 340:359–368
Tomita M, Kawakami H, Uchihara JN, Okudaira T, Masuda M, Matsuda T et al (2006) Inhibition of constitutively active Jak-Stat pathway suppresses cell growth of human T-cell leukemia virus type 1-infected T-cell lines and primary adult T-cell leukemia cells. Retrovirology 3:22
Tomita M, Kawakami H, Uchihara JN, Okudaira T, Masuda M, Takasu N et al (2006) Curcumin (diferuloylmethane) inhibits constitutive active NF-kappaB, leading to suppression of cell growth of human T-cell leukemia virus type I-infected T-cell lines and primary adult T-cell leukemia cells. Int J Cancer 118:765–772
Tomita M, Matsuda T, Kawakami H, Uchihara JN, Okudaira T, Masuda M et al (2006) Curcumin targets Akt cell survival signaling pathway in HTLV-I-infected T-cell lines. Cancer Sci 97:322–327
Kirken RA, Erwin RA, Wang L, Wang Y, Rui H, Farrar WL (2000) Functional uncoupling of the Janus kinase 3-Stat5 pathway in malignant growth of human T cell leukemia virus type 1-transformed human T cells. J Immunol 165:5097–5104
Kawakami H, Tomita M, Okudaira T, Ishikawa C, Matsuda T, Tanaka Y et al (2007) Inhibition of heat shock protein-90 modulates multiple functions required for survival of human T-cell leukemia virus type I-infected T-cell lines and adult T-cell leukemia cells. Int J Cancer 120:1811–1820
Banerji U, O’Donnell A, Scurr M, Pacey S, Stapleton S, Asad Y et al (2005) Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol 23:4152–4161
Yonehara S, Ishii A, Yonehara M (1989) A cell-killing monoclonal antibody (anti-Fas) to a cell surface antigen co-downregulated with the receptor of tumor necrosis factor. J Exp Med 169:1747–1756
Debatin KM, Goldman CK, Waldmann TA, Krammer PH (1993) APO-1-induced apoptosis of leukemia cells from patients with adult T-cell leukemia. Blood 81:2972–2977
Debatin KM, Goldmann CK, Bamford R, Waldmann TA, Krammer PH (1990) Monoclonal-antibody-mediated apoptosis in adult T-cell leukaemia. Lancet 335:497–500
Waldmann TA, White JD, Goldman CK, Top L, Grant A, Bamford R et al (1993) The interleukin-2 receptor: a target for monoclonal antibody treatment of human T-cell lymphotrophic virus I-induced adult T-cell leukemia. Blood 82:1701–1712
Waldmann TA, Goldman CK, Bongiovanni KF, Sharrow SO, Davey MP, Cease KB et al (1988) Therapy of patients with human T-cell lymphotrophic virus I-induced adult T-cell leukemia with anti-Tac, a monoclonal antibody to the receptor for interleukin-2. Blood 72:1805–1816
Callens C, Moura IC, Lepelletier Y, Coulon S, Renand A, Dussiot M et al (2008) Recent advances in adult T-cell leukemia therapy: focus on a new anti-transferrin receptor monoclonal antibody. Leukemia 22:42–48
Waldmann TA, White JD, Carrasquillo JA, Reynolds JC, Paik CH, Gansow OA et al (1995) Radioimmunotherapy of interleukin-2R alpha-expressing adult T-cell leukemia with Yttrium-90-labeled anti-Tac. Blood 86:4063–4075
Waldmann TA (2007) Daclizumab (anti-Tac, Zenapax) in the treatment of leukemia/lymphoma. Oncogene 26:3699–3703
Phillips KE, Herring B, Wilson LA, Rickford MS, Zhang M, Goldman CK et al (2000) IL-2Ralpha-Directed monoclonal antibodies provide effective therapy in a murine model of adult T-cell leukemia by a mechanism other than blockade of IL-2/IL-2Ralpha interaction. Cancer Res 60:6977–6984
Tan C, Waldmann TA (2002) Proteasome inhibitor PS-341, a potential therapeutic agent for adult T-cell leukemia. Cancer Res 62:1083–1086
Zhang Z, Zhang M, Goldman CK, Ravetch JV, Waldmann TA (2003) Effective therapy for a murine model of adult T-cell leukemia with the humanized anti-CD52 monoclonal antibody, Campath-1H. Cancer Res 63:6453–6457
Moura IC, Lepelletier Y, Arnulf B, England P, Baude C, Beaumont C et al (2004) A neutralizing monoclonal antibody (mAb A24) directed against the transferrin receptor induces apoptosis of tumor T lymphocytes from ATL patients. Blood 103:1838–1845
Vidal C, Matsushita S, Colamonici OR, Trepel JB, Mitsuya H, Neckers LM (1988) Human T lymphotropic virus I infection deregulates surface expression of the transferrin receptor. J Immunol 141:984–988
Maeda Y, Miyatake J, Sono H, Matsuda M, Tatsumi Y, Horiuchi F et al (1996) 13-cis retinoic acid inhibits growth of adult T cell leukemia cells and causes apoptosis; possible new indication for retinoid therapy. Intern Med 35:180–184
Fujimura S, Suzumiya J, Anzai K, Ohkubo K, Hata T, Yamada Y et al (1998) Retinoic acids induce growth inhibition and apoptosis in adult T-cell leukemia (ATL) cell lines. Leuk Res 22:611–618
Furuke K, Sasada T, Ueda-Taniguchi Y, Yamauchi A, Inamoto T, Yamaoka Y et al (1997) Role of intracellular redox status in apoptosis induction of human T-cell leukemia virus type I-infected lymphocytes by 13-cis-retinoic acid. Cancer Res 57:4916–4923
Darwiche N, Hatoum A, Dbaibo G, Kadara H, Nasr R, Abou-Lteif G et al (2004) N-(4-hydroxyphenyl)retinamide induces growth arrest and apoptosis in HTLV-I-transformed cells. Leukemia 18:607–615
Boya P, Morales MC, Gonzalez-Polo RA, Andreau K, Gourdier I, Perfettini JL et al (2003) The chemopreventive agent N-(4-hydroxyphenyl)retinamide induces apoptosis through a mitochondrial pathway regulated by proteins from the Bcl-2 family. Oncogene 22:6220–6230
Darwiche N, Abou-Lteif G, Bazarbachi A (2007) Reactive oxygen species mediate N-(4-hydroxyphenyl)retinamide-induced cell death in malignant T cells and are inhibited by the HTLV-I oncoprotein Tax. Leukemia 21:261–269
Maeda Y, Yamaguchi T, Ueda S, Miyazato H, Matsuda M, Kanamaru A (2004) All-trans retinoic acid reduced skin involvement of adult T-cell leukemia. Leukemia 18:1159–1160
Inozume T, Matsue H, Furuhashi M, Nakamura Y, Mitsui H, Ando N et al (2005) Successful use of etretinate for long-term management of a patient with cutaneous-type adult T-cell leukaemia/lymphoma. Br J Dermatol 153:1239–1241
Nawata H, Maeda Y, Sumimoto Y, Miyatake J, Kanamaru A (2001) A mechanism of apoptosis induced by all-trans retinoic acid on adult T-cell leukemia cells: a possible involvement of the Tax/NF-kappaB signaling pathway. Leuk Res 25:323–331
Darwiche N, El-Sabban M, Bazzi R, Nasr R, Al-Hashimi S, Hermine O et al (2001) Retinoic acid dramatically enhances the arsenic trioxide-induced cell cycle arrest and apoptosis in retinoic acid receptor alpha-positive human T-cell lymphotropic virus type-I-transformed cells. Hematol J 2:127–135
Gill PS, Harrington W Jr, Kaplan MH, Ribeiro RC, Bennett JM, Liebman HA et al (1995) Treatment of adult T-cell leukemia-lymphoma with a combination of interferon alfa and zidovudine. N Engl J Med 332:1744–1748
Hermine O, Bouscary D, Gessain A, Turlure P, Leblond V, Franck N et al (1995) Brief report: treatment of adult T-cell leukemia-lymphoma with zidovudine and interferon alfa. N Engl J Med 332:1749–1751
Bazarbachi A, Nasr R, El-Sabban ME, Mahe A, Mahieux R, Gessain A et al (2000) Evidence against a direct cytotoxic effect of alpha interferon and zidovudine in HTLV-I associated adult T cell leukemia/lymphoma. Leukemia 14:716–721
Datta A, Nicot C (2007) Telomere attrition induces a DNA double-strand break damage signal that reactivates p53 transcription in HTLV-I leukemic cells. Oncogene 27:1135–1141
Datta A, Bellon M, Sinha-Datta U, Bazarbachi A, Lepelletier Y, Canioni D et al (2006) Persistent inhibition of telomerase reprograms adult T-cell leukemia to p53-dependent senescence. Blood 108:1021–1029
Ikeda K, Oka M, Yamada Y, Soda H, Fukuda M, Kinoshita A et al (1999) Adult T-cell leukemia cells over-express the multidrug-resistance-protein (MRP) and lung-resistance-protein (LRP) genes. Int J Cancer 82:599–604
Yasunami T, Wang YH, Tsuji K, Takanashi M, Yamada Y, Motoji T (2007) Multidrug resistance protein expression of adult T-cell leukemia/lymphoma. Leuk Res 31:465–470
Lau A, Nightingale S, Taylor GP, Gant TW, Cann AJ (1998) Enhanced MDR1 gene expression in human T-cell leukemia virus-I-infected patients offers new prospects for therapy. Blood 91:2467–2474
Hamamura RS, Ohyashiki JH, Kurashina R, Kobayashi C, Zhang Y, Takaku T et al (2007) Induction of heme oxygenase-1 by cobalt protoporphyrin enhances the antitumour effect of bortezomib in adult T-cell leukaemia cells. Br J Cancer 97:1099–1105
Okudaira T, Tomita M, Uchihara JN, Matsuda T, Ishikawa C, Kawakami H et al (2006) NIK-333 inhibits growth of human T-cell leukemia virus type I-infected T-cell lines and adult T-cell leukemia cells in association with blockade of nuclear factor-kappaB signal pathway. Mol Cancer Ther 5:704–712
Watanabe M, Ohsugi T, Shoda M, Ishida T, Aizawa S, Maruyama-Nagai M et al (2005) Dual targeting of transformed and untransformed HTLV-1-infected T cells by DHMEQ, a potent and selective inhibitor of NF-kappaB, as a strategy for chemoprevention and therapy of adult T-cell leukemia. Blood 106:2462–2471
Ariga A, Namekawa J, Matsumoto N, Inoue J, Umezawa K (2002) Inhibition of tumor necrosis factor-alpha-induced nuclear translocation and activation of NF-kappa B by dehydroxymethylepoxyquinomicin. J Biol Chem 277:24625–24630
Okudaira T, Hirashima M, Ishikawa C, Makishi S, Tomita M, Matsuda T et al (2007) A modified version of galectin-9 suppresses cell growth and induces apoptosis of human T-cell leukemia virus type I-infected T-cell lines. Int J Cancer 120:2251–2261
Mori N, Matsuda T, Tadano M, Kinjo T, Yamada Y, Tsukasaki K et al (2004) Apoptosis induced by the histone deacetylase inhibitor FR901228 in human T-cell leukemia virus type 1-infected T-cell lines and primary adult T-cell leukemia cells. J Virol 78:4582–4590
Zhang J, Nagasaki M, Tanaka Y, Morikawa S (2003) Capsaicin inhibits growth of adult T-cell leukemia cells. Leuk Res 27:275–283
Harakeh S, Diab-Assaf M, Abu-El-Ardat K, Niedzwiecki A, Rath M (2006) Mechanistic aspects of apoptosis induction by l-lysine in both HTLV-1-positive and -negative cell lines. Chem Biol Interact 164:102–114
Ishitsuka K, Ikeda R, Utsunomiya A, Uozumi K, Hanada S, Suzuki S et al (2002) Arsenic trioxide induces apoptosis in HTLV-I infected T-cell lines and fresh adult T-cell leukemia cells through CD95 or tumor necrosis factor alpha receptor independent caspase activation. Leuk Lymphoma 43:1107–1114
Dasgupta A, Jung KJ, Jeong SJ, Brady JN (2008) Inhibition of methyltransferases results in induction of g2/m checkpoint and programmed cell death in human T-lymphotropic virus type 1-transformed cells. J Virol 82:49–59
Harakeh S, Diab-Assaf M, Khalife JC, Abu-el-Ardat KA, Baydoun E, Niedzwiecki A et al (2007) Ascorbic acid induces apoptosis in adult T-cell leukemia. Anticancer Res 27:289–298
Jeong SJ, Dasgupta A, Jung KJ, Um JH, Burke A, Park HU et al (2008) PI3K/AKT inhibition induces caspase-dependent apoptosis in HTLV-1-transformed cells. Virology 370:264–272
Harakeh S, Abu-El-Ardat K, Diab-Assaf M, Niedzwiecki A, El-Sabban M, Rath M (2008) Epigallocatechin-3-gallate induces apoptosis and cell cycle arrest in HTLV-1-positive and -negative leukemia cells. Med Oncol 25:30–39
Moarbess G, El-Hajj H, Kfoury Y, El-Sabban ME, Lepelletier Y, Hermine O et al (2008) EAPB0203, a member of the imidazoquinoxaline family, inhibits growth and induces caspase dependent apoptosis in T cell lymphomas and HTLV-I associated adult T-cell leukemia/lymphoma. Blood 111:3770–3777
Haneji K, Matsuda T, Tomita M, Kawakami H, Ohshiro K, Uchihara JN et al (2005) Fucoidan extracted from Cladosiphon okamuranus Tokida induces apoptosis of human T-cell leukemia virus type 1-infected T-cell lines and primary adult T-cell leukemia cells. Nutr Cancer 52:189–201
Hayashibara T, Yamada Y, Nakayama S, Harasawa H, Tsuruda K, Sugahara K et al (2002) Resveratrol induces downregulation in survivin expression and apoptosis in HTLV-1-infected cell lines: a prospective agent for adult T cell leukemia chemotherapy. Nutr Cancer 44:193–201
Harakeh S, Diab-Assef M, El-Sabban M, Haddadin M, Gali-Muhtasib H (2004) Inhibition of proliferation and induction of apoptosis by 2-benzoyl-3-phenyl-6,7-dichloroquinoxaline 1,4-dioxide in adult T-cell leukemia cells. Chem Biol Interact 148:101–113
Achachi A, Florins A, Gillet N, Debacq C, Urbain P, Foutsop GM et al (2005) Valproate activates bovine leukemia virus gene expression, triggers apoptosis, and induces leukemia/lymphoma regression in vivo. Proc Natl Acad Sci USA 102:10309–10314
Lezin A, Gillet N, Olindo S, Signate A, Grandvaux N, Verlaeten O et al (2007) Histone deacetylase mediated transcriptional activation reduces proviral loads in HTLV-1 associated myelopathy/tropical spastic paraparesis patients. Blood 110:3722–3728
Nishioka C, Ikezoe T, Yang J, Komatsu N, Bandobashi K, Taniguchi A et al (2008) Histone deacetylase inhibitors induce growth arrest and apoptosis of HTLV-1-infected T-cells via blockade of signaling by nuclear factor kappaB. Leuk Res 32:287–296
Hasegawa H, Yamada Y, Komiyama K, Hayashi M, Ishibashi M, Yoshida T et al (2006) Dihydroflavonol BB-1, an extract of natural plant Blumea balsamifera, abrogates TRAIL resistance in leukemia cells. Blood 107:679–688
Sinha-Datta U, Taylor JM, Brown M, Nicot C (2008) Celecoxib disrupts the canonical apoptotic network in HTLV-I cells through activation of Bax and inhibition of PKB/Akt. Apoptosis 13:33–40
Harakeh S, Diab-Assaf M, Niedzwiecki A, Khalife J, Abu-El-Ardat K, Rath M (2006) Apoptosis induction by Epican Forte in HTLV-1 positive and negative malignant T-cells. Leuk Res 30:869–881
Waldmann TA, Longo DL, Leonard WJ, Depper JM, Thompson CB, Kronke M et al (1985) Interleukin 2 receptor (Tac antigen) expression in HTLV-I-associated adult T-cell leukemia. Cancer Res 45:4559s–4562s
Acknowledgments
The authors wish to thank Dr. V. Ciminale (Department of Oncology and Surgical Sciences, University of Padua, Italy); Dr. J. Semmes (Department of Microbiology and Molecular Cell Biology, Eastern Virginia Medical School, Norfolk, Virginia 23507, USA); Dr. J.M. Mesnard (Laboratoire Infections Rétrovirales et Signalisation Cellulaire, CNRS/UM I UMR 5121/IFR 122, Institut de Biologie, 34000 Montpellier, France) for kindly providing pictures for cellular localization of p13, Tax and HBZ, respectively. This work was supported by grants CA106258 and CA115398 from the National cancer Institute to C. Nicot.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Taylor, J.M., Nicot, C. HTLV-1 and apoptosis: role in cellular transformation and recent advances in therapeutic approaches. Apoptosis 13, 733–747 (2008). https://doi.org/10.1007/s10495-008-0208-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-008-0208-7