
Abstract
Adult T-cell leukemia/lymphoma (ATLL) is an aggressive lymphoproliferative disease of very poor clinical prognosis associated with infection by the human T-cell leukemia virus type I (HTLV-I). Treatment of patients with ATLL using conventional chemotherapy has limited benefit because HTLV-I cells are refractory to most apoptosis-inducing agents. In this study, we report that Celecoxib induces cell death via the intrinsic mitochondrial pathway in HTLV-I transformed leukemia cells. Treatment with Celecoxib was associated with activation of Bax, decreased expression of Mcl-1, loss of the mitochondrial membrane potential and caspase-9-dependent apoptosis. These effects were independent from Bcl-2 and Bcl-xL. We also found that Celecoxib inhibited the Akt/GSK3 β survival pathway in HTLV-I cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adult T-cell leukemia (ATL) is etiologically linked to the human T-cell leukemia virus type I (HTLV-I) [1]. HTLV-I-mediated T-cell transformation arises from a multi-step oncogenic process in which the virus induces chronic proliferation of the infected T-cells. The mechanism by which HTLV-I engenders ATL is not clear, but the long latency period of several decades preceding the disease suggests it relies upon long term survival, proliferation and avoidance of telomere shortening of virus-infected cells [2–5]. Though many aspects of HTLV-I biology have been uncovered, the treatment of the disease remains disappointing, with minimal improvement in the overall survival of infected patients [6]. Some success has been reported in treating ATL with a combination of zidovudine (AZT) and IFN-α [7]. However, this success is fleeting as many patients relapse, because response to therapy requires the presence of a transcriptionally active p53 gene [8]. Other treatments including bone marrow transplantation, topoisomerase inhibitors, anti-Tac monoclonal antibody, inhibitors of NF-kB and proteasome inhibitors have been previously reviewed [9]. Much conventional chemotherapy has limited benefits since HTLV-I cells are resistant to conventional anti-cancer, apoptosis inducing agents in part through upregulated expression of Bcl-xL in ATL patients [10].
The cyclooxygenase (Cox) family of enzymes has been implicated in the process of cell proliferation and angiogenesis for many tumors, including colon, non-small-cell lung, breast, gastric, esophagus, prostrate, head/neck and cholangiocarcinoma [11, 12]. Cox-2, an immediate early gene that is induced by a variety of stimuli, such as cytokines, hormones, mitogens and growth factors, has been reported to be associated with tumor development and progression, as well as to protect cells from apoptosis induced by various cellular stresses. Elevated Cox-2 expression has been observed in various types of cancers including HTLV-I associated ATL [13]. Cyclooxygenase-2 has proved to be an important cellular target for both therapy and or prevention of inflammatory disorders and cancers. Celecoxib, a selective cyclooxegenase-2 (Cox-2) inhibitor is a non steroidal anti-inflammatory (NSAIDs) drug and is known to have anti cancer effects [14–16]. The tumor suppression activity of Celecoxib has been related to induction of apoptosis in many cancer cell lines. While Celecoxib is a specific inhibitor of cox-2, recent data indicate that its apoptotic properties may be mediated through a cox-2 independent pathway inasmuch as Jurkat T-cells, which do not express cox-2, are sensitive to Celecoxib (data not shown) [17].
In this study, we report that Celecoxib has a potent anti-proliferative effect on HTLV-I infected cells in vitro. Celecoxib induced conformational change and activation of Bax, PARP cleavage, caspase-9 activation and apoptosis. In addition, we found that Celecoxib mediated anti-proliferation involved disruption of Akt signaling resulting in activation of GSK3β.
Experimental procedures
Cell lines and Cox-2 inhibitor
MT-2 and C8166, HTLV-I-transformed T-cell lines was grown in RPMI-1640 (Invitrogen Carlsbad, CA) supplemented with 10% fetal bovine serum, gentamycin, and penicillin-streptomycin. 1185, LAF and SP, IL-2 dependent T-cell lines immortalized by HTLV-I, and peripheral blood mononuclear cells (PBMCs) were cultured in 20% serum and 40 U/ml IL-2. Celecoxib (SC-58635), Cox-2 inhibitor, was obtained from Pfizer and it was dissolved in DMSO and used at concentrations 20–100 μM.
Cell-proliferation assay
Cell proliferation was measured using the Cell Proliferation Kit II (XTT; Roche, Mannheim, Germany) according to manufacturer’s instructions. Cells (105/ml) were treated with cox-2 inhibitor (concentrations ranging from 0 to 100 μM) for 24 h in a 96-well plate and then incubated with tetrazolium salt XTT for 4 h. Proliferation was quantified by measuring cleavage of XTT to an orange formazan dye using an enzyme-linked immuno-absorbent assay (ELISA) reader at 450 nm. Assays were performed in duplicates.
Apoptosis assay
Cells (5 × 105) were treated with cox-2 inhibitor (concentrations ranging from 0 to 100 μM) for 24 h and then collected and washed with cold PBS. Cells were then stained with Annexin V/propidium iodide using the Vybrant Apoptosis Assay Kit no. 2 (Molecular Probes, Eugene, OR) according to the manufacturer’s instructions.
Mitochondrial membrane potential
Cells (1 × 106) were treated with 80 μM Celecoxib for 24 h, then change in the mitochondrial membrane potential (ΔΨm) was measured using the ApoAlert Mitochondrial Membrane Sensor Kit (Clontech, Mountain View, CA) according to the manufacturer’s instructions.
Immunoprecipitation of activated Bax
Cells were harvested in either 2% (w/v) CHAPS or 1% (v/v) Triton-X-100 lysis buffer containing 137 mM NaCl, 20 mM Tris (pH 7.4), aprotonin, leupeptin, and pepstatin. Cells were lysed for one hour at 4°C, and supernatants were incubated with anti-Bax(N20) overnight. Immune complexes were isolated using proteinA/G sepharose (Santa Cruz Biotechnology), and purified proteins were released in SDS-PAGE sample buffer.
Western blot analysis
Cells were washed with PBS containing phosphatase inhibitors and lysed in RIPA buffer with phosphatase and protease inhibitors (Complete Cocktail; Roche). Protein concentration was determined with the Bio-Rad protein assay (Bio-Rad, Hercules, CA). 30 μg of protein lysates were separated on 8–15% SDS–polyacrylamide gel electrophoresis and transferred to Immobilon PVDF membranes (Millipore, Billerica, MA). Proteins were detected with the appropriate primary antibody [Anti-phospho-AKT(Ser473) (Cell Signaling), anti-phospho-GSK-3β(Ser9) (Cell Signaling), anti-Bax(N20) (Santa Cruz Biotechnology), anti-Mcl-1 (Santa Cruz Biotechnology), anti-Bcl-xL, anti-Bcl-2, anti-Bak, anti-PARP, anti-AKT (Cell Signaling), anti-GSK-3β, anti-actin (Santa Cruz Biotechnology)] followed by an anti-rabbit or anti-mouse IgG-HRP-conjugated donkey antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Blots were developed using a chemiluminescent detection system (Pierce).
Results
Anti-proliferative effect of Celecoxib (Cox-2 inhibitor) on HTLV-I infected cells
Since Celecoxib has been used to inhibit cancer cell growth and target tumor cells to apoptosis we tested its effect on HTLV-I leukemic cells. Primary blood mononuclear cells (PBMCs) were used as a control to verify the selective toxicity of Celecoxib towards cancer cells at doses used in this study. Cells were plated at the same density and treated with increasing dose of Celecoxib. Cellular proliferation was assessed using the Cell Proliferation Kit II XTT as previously reported [18]. Proliferation was quantified by measuring cleavage of XTT to an orange formazan dye using an ELISA reader at 450 nm. Results are mean values of at least two independent experiments each performed in duplicate. Our data indicated that Celecoxib, at 24 h, has a dose dependent anti-proliferative effect on HTLV-I transformed cells, MT2 (Fig. 1a). In contrast Celecoxib did not have a significant effect on normal PBMC (Fig. 1a). We next tested three additional HTLV-I infected cell lines (1185, LAF and SP) and found that they were similarly sensitive to Celecoxib (Fig. 1b) when compared to PBMC, suggesting that, in general, HTLV-I infected cells are susceptible to the Cox-2 inhibitor, Celecoxib.

Celecoxib has anti-proliferative effect on HTLV-I infected cells. (a) Proliferation assay of healthy PBMC and HTLV-I infected cells (MT2) treated with increasing concentrations of Celecoxib at 24 h. (b) Comparison of PBMC, MT2, LAF, 1185 and SP proliferation following treatment with 80 μM Celecoxib
Celecoxib targets the HTLV-I infected cells towards apoptosis
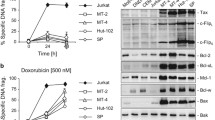
To gain insight into the cellular fate induced by Celecoxib, we analyzed treated cells by flow cytometry using double staining (Annexin V/PI). Apoptotic cells are scored as Annexin V positive and PI negative whereas necrotic cells are Annexin V negative and PI positive. Our results indicate that Celecoxib triggers apoptosis in MT2 cells in a dose dependent manner which is shown by the dose dependent increase in positive Annexin V staining (Fig. 2a). In contrast Celecoxib-treated PBMC did not show any positive Annexin V staining (Fig. 2a). We then examined expression/regulation of downstream apoptotic markers such as caspases-8, 9 and PARP. Caspase-9 is activated immediately downstream of the mitochondria following cytochrome c release and apoptosome formation. It has been previously reported that apoptosis induced by Celecoxib is mediated by caspases-9 and 8 was not required [19]. Cell lysates from both MT2 and C8166, HTLV-1-infected cells, exhibited cleaved caspase-9 when treated with 100 μM of Celecoxib (Fig. 2b), a dose similar to that used in other studies. In contrast, caspase-8, traditionally activated by death receptor signaling, was not activated by Celecoxib in HTLV-I treated cells. We also assessed the cleavage of the apoptotic substrate PARP by western blotting (Fig. 2c). Untreated MT2 or C8166 cells showed no detectable levels of PARP cleavage. Western blotting of lysates from both cell lines, however, clearly demonstrated the appearance of cleaved PARP at higher Celecoxib concentrations (Fig. 2c). The fact that apoptosis was detected by Annexin V staining at lower doses of Celecoxib is consistent with other reports and may be related to the fact that Annexin V is an early marker of apoptosis, and, to the relative sensitivity of each assay. Together our data indicates that Celecoxib-treated HTLV-1 infected cells undergo apoptosis. We also assessed the expression levels of other proteins involved in the apoptotic cascade. We found high levels of Mcl-1 expression in HTLV-I transformed cells which decreased significantly in MT2 and C8166 HTLV-1-infected cell lines treated with increasing concentrations of Celecoxib (Fig. 2d). Intriguingly, we also noted a significant increase in both Bax and Bcl-xL in MT2 cells and C8166 treated cells (Fig. 2d). Although Bcl-xL is an important anti-apoptotic factor in HTLV-I cells, the surge in levels of Bax expression cannot be counteracted by high levels of Bcl-xL, since the latter does not prevent Bax-mediated apoptosis [20]. In contrast, Bcl-2 levels were unchanged (Fig. 2d).

Celecoxib treatment of HTLV-1-infected cell lines induces apoptosis. (a) MT2 cells following treatment with 0–100 μM Celecoxib, were harvested, washed in PBS without Ca2+ and Mg2+ and stained with Vybrant Apoptosis kit. Annexin V conjugated to flourescein allowed the identification of apoptosis (AV+/PI−) by flourescein activated cell sorting (FACS). (b) Western blot analysis of caspases-8 and 9 in HTLV-I infected cells (MT2 and C8166) following treatment with increasing amounts of Celecoxib. (c) Western blot analysis of PARP cleavage in MT2 and C8166 cells following treatment with increasing concentrations of Celecoxib. (d) Western blot analysis of Mcl-1, Bax, Bcl-xL and Bcl-2 in Celecoxib 0–100 μM treated MT2 and C8166 cells
Celecoxib induces loss of membrane potential involving Bax activation in HTLV-I cells
The mitochondria are a critical checkpoint in the intrinsic apoptotic cascade. Changes in the mitochondrial membrane potential, ΔΨm, a well characterized marker of cells undergoing apoptosis, were measured following Celecoxib treatment of MT2 cells (Fig. 3a). MT2 cells treated with Celecoxib showed a collapse in the ΔΨm indicating that these cells underwent apoptosis following Celecoxib treatment (Fig. 3a). In contrast, PBMCs treated with Celecoxib did not show any alteration of the membrane potential. Apoptosis initiated through the mitochondria typically involves the activation of the pro-apoptotic multi-domain Bcl-2 family member Bax. We further assessed whether the observed apoptotic phenotype of HTLV-1-infected cells treated with Celecoxib involved the mitochondrial pathway by assessing Bax activation. During apoptosis Bax undergoes a conformational change that reveals an N-terminal epitope [21, 22]. PBMC, MT2 and C8166 cells were treated with increasing concentrations of Celecoxib and were lysed in CHAPS lysis buffer to maintain the conformation of Bax. Activated Bax was then immunoprecipitated using an N-terminal conformation specific anti-Bax antibody, and immunoprecipitates were blotted for Bax. Healthy cells displayed no active Bax as assessed by immunoprecipitation, while cells lysed in Triton-X-100 as control, which unfolds Bax into an activated form, did display activated Bax (Fig. 3b). MT2 cells and C8166 cells, but not PBMC, showed a dose-dependent increase in active Bax following treatment with Celecoxib (Fig. 3b), indicating that Celecoxib induces apoptotic signaling events that involve Bax activation and the mitochondrial intrinsic pathway.

Celecoxib treatment triggers loss of membrane potential and Bax activation. (a) ΔΨm collapse was measured following treatment of PBMC and MT2 cells with 80 μM Celecoxib using the Apoalert Mitochondrial Membrane Sensor kit. Results are representative of at least two experiments. (b) PBMC, MT2 and C8166 cells were treated with the indicated concentrations of Celecoxib, lysed in 2% CHAPS or 1% Triton-X-100 lysis buffer, and immuno-precipitated with anti-Bax (N20). Immuno-precipitates and lysates were immuno-blotted with anti-Bax
Celecoxib disrupts Akt signaling in HTLV-I infected cells
Activation of the PI3K/Akt pathway has been associated with malignant transformation [23]. Recently, several targets of the PI3K/Akt pathway have been identified which contribute to its promotion of cell survival. Among these are BAD, caspase-9, the forkhead family, and the NF-kB transcription factor [24]. Activation of the PI3K/Akt survival pathway is critical for proliferation of HTLV-I transformed cells [25, 26]. Several studies have shown that treatment with drugs that disrupt this pathway lead to growth arrest and apoptosis. In cell lines derived in vitro, Tax stimulates Akt resulting in phosphorylation and inactivation of GSK3β and the constitutive activation of β-catenin [27]. In addition, the viral protein p30 has also been shown to potently inhibit GSK3β and likely contributes to the activation of the β-catenin/Wnt pathway in HTLV-I infected cells [28]. The activation and phosphorylation of the signaling molecules AKT and GSK-3β in Celecoxib treated cells was assessed by western blotting. As seen in Fig. 4, C8166 cells treated with increasing concentrations of Celecoxib exhibited a loss of phosphorylated AKT, suggesting that AKT is de-activated following treatment. Consistent with these results, we found a decrease in the phosphorylation of GSK-3β in C8166 cells following treatment with Celecoxib. Reactivation of GSK3β likely inhibits β-catenin. Together our data suggest that Celecoxib targets active Bax to the mitochondria and inhibits the Akt/GSK3β/β-catenin pathway leading to apoptotic death of HTLV-I leukemic cells.

Celecoxib disrupts AKT signaling in HTLV-I infected cells. (a) Western blot analysis of Phoso AKT, AKT, Phospho GSK3β and GSK3β in C8166 cells following Celecoxib treatment for 48 h. The numbers below represent quantitative analysis of the bands. (b) Quantitative analysis of the decrease in phospho-AKT and phospho-GSK3β relative to total AKT and total GSK3β, respectively, in the cells
Discussion
Adult T-cell leukemia has a poor clinical prognosis and treatment of patients using conventional chemotherapy has limited benefit given that HTLV-I cells are resistant to conventional anti-cancer, apoptosis-inducing agents. In this study we found a potent anti-proliferative effect of Celecoxib against HTLV-I transformed cells at doses that do not affect primary T-cells. These effects were time and dose dependent and similarly affected both IL-2-dependent or IL-2-independent T-cells infected by HTLV-I. Anti-proliferative effects were associated with apoptosis as shown by Annexin V staining and the activation of caspase-9. We found following treatment with Celecoxib, Bax undergoes a conformational change that reveals an N-terminal epitope which can be detected by immunoprecipitation with an N-terminal conformation-specific antibody. This activated form of Bax is known to relocate to the mitochondria and induces the loss of the mitochondrial membrane potential, leading to release of cytochrome c and activation of caspases.
Anti-apoptotic Bcl-2 family members protect mitochondria from pro-apoptotic stimuli. In this study we observed a loss of the mitochondrial membrane potential in HTLV-I cells following Celecoxib treatment. Altogether, this supports the notion that Celecoxib induces cell death via the intrinsic mitochondrial pathway. HTLV-1-infected cells treated with increasing concentrations of Celecoxib exhibited a significant loss of Mcl-1 expression. Mcl-1 normally complexes with pro-apoptotic proteins such as Bak and Bim at the mitochondria [29, 30], and the loss of Mcl-1 releases these pro-apoptotic factors, which are then able to induce apoptosis through the mitochondrial pathway. Celecoxib treatment has been shown to also induce the loss of Mcl-1 in hepatocellular carcinoma cells, and this is followed by Bax activation and translocation to the mitochondria [31]. It is interesting to speculate that reduced Mcl-1 expression induced by Celecoxib may free pro-apoptotic Bcl-2 family members such as Bak or Bim, leading to apoptosis. Whether the loss of Mcl-1 is required for Celecoxib-induced death, however, remains to be investigated. Mcl-1 is continuously synthesized, ubiquitinated and degraded [32]. Stimuli which turn off Mcl-1 transcription, such as DNA damaging agents, downregulate Mcl-1 transcription; since no new Mcl-1 is produced, a loss of existing Mcl-1 is thus observed. Whether treatment with Celecoxib affects mcl-1 promoter expression warrants future studies. Paradoxically, we observed an increase in Bcl-xL expression in HTLV-I-infected cells following Celecoxib treatment. This is similar to other observations where treatment of lung cancer cells with Celecoxib induces an NF-κB response, resulting in Bcl-xL upregulation [33]. As mentioned above Bcl-xL, a pro-survival member of the Bcl-2 family, does not have the capacity to directly inhibit Bax, and instead specifically inhibits certain upstream BH3-only proteins. This supports our observation that the upregulation of Bcl-xL in C8166 cells is unable to inhibit Celecoxib-induced apoptosis and Bax activation, leading to death of the cell.
Whether the overexpression of either Bcl-2, Mcl-1, or other anti-apoptotic proteins can protect Celecoxib-treated HTLV-1 cell lines remains to be seen. Indeed, the overexpression of Mcl-1 protects hepatocellular carcinoma cells from Celecoxib-induced death, suggesting that particular Bcl-2 family proteins can inhibit Celecoxib-induced death, and that specific BH3-only proteins are likely activated by Celecoxib to trigger apoptosis. It is possible that the loss of Mcl-1 is an important step in Celecoxib-induced apoptosis in HTLV-1-infected cells.
Previous reports have indicated that Celecoxib can influence upstream kinase signaling pathways to promote cell death. In order to address the mechanism used by Celecoxib to induce apoptosis in HTLV-1-infected cells, we examined upstream kinase signaling pathway Akt, a major pro-survival signaling protein which up-regulates expression of number of proteins involved in cell survival. Several studies have highlighted the importance of Akt activation for the survival and growth of HTLV-I tumor cells. Indeed, we and others found that treatment with Celecoxib resulted in the significant loss of phosphorylated and unphosphorylated Akt [34]. Since active Akt induces the phosphorylation and subsequent inactivation of GSK3β at Ser9, we assessed the level of GSK3β phosphorylation following Celecoxib treatment. HTLV-I-infected cells treated with increasing concentrations of Celecoxib resulted in the remarkable decrease in phosphorylated GSK3β, indicative of an increase in activated GSK3β. Active GSK3β has the capacity to phosphorylate and inactivate downstream protein targets such as β-catenin, which normally regulates the Wnt signaling pathway controlling cell survival. Our results suggest that Celecoxib targets Akt/GSK3β pathway. In conclusion, our results confirm that although multiple apoptotic pathways are inhibited in HTLV-1-infected cells, these effects are reversible and targeting these pathways may open new avenues for treating HTLV-1-infected patients.
References
Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC (1980) Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci USA 77:7415–7419
Franchini G, Nicot C, Johnson JM (2003) Seizing of T cells by human T-cell leukemia/lymphoma virus type 1. Adv Cancer Res 89:69–132
Bellon M, Datta A, Brown M et al (2006) Increased expression of telomere length regulating factors TRF1, TRF2 and TIN2 in patients with adult T-cell leukemia. Int J Cancer 119:2090–2097
Sinha-Datta U, Horikawa I, Michishita E et al (2004) Transcriptional activation of hTERT through the NF-κB pathway in HTLV-I-transformed cells. Blood 104:2523–2531
Grassmann R, Aboud M, Jeang KT (2005) Molecular mechanisms of cellular transformation by HTLV-1 Tax. Oncogene 24:5976–5985
Bazarbachi A, Ghez D, Lepelletier Y et al (2004) New therapeutic approaches for adult T-cell leukaemia. Lancet Oncol 5:664–672
Hermine O, Allard I, Levy V, Arnulf B, Gessain A, Bazarbachi A (2002) A prospective phase II clinical trial with the use of zidovudine and interferon-α in the acute and lymphoma forms of adult T-cell leukemia/lymphoma. Hematol J 3:276–282
Datta A, Bellon M, Sinha-Datta U et al (2006) Persistent inhibition of telomerase reprograms adult T-cell leukemia to p53-dependent senescence. Blood 108:1021–1029
Bazarbachi A, Hermine O (2001) Treatment of adult T-cell leukaemia/lymphoma: current strategy and future perspectives. Virus Res 78:79–92
Nicot C, Mahieux R, Takemoto S, Franchini G (2000) Bcl-X(L) is up-regulated by HTLV-I and HTLV-II in vitro and in ex vivo ATLL samples. Blood 96:275–281
Hwang D, Scollard D, Byrne J, Levine E (1998) Expression of cyclooxygenase-1 and cyclooxygenase-2 in human breast cancer. J Natl Cancer Inst 90:455–460
Soslow RA, Dannenberg AJ, Rush D et al (2000) COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer 89:2637–2645
Mori N, Inoue H, Yoshida T, Tanabe T, Yamamoto N (2001) Constitutive expression of the cyclooxygenase-2 gene in T-cell lines infected with human T cell leukemia virus type I. Int J Cancer 94:813–819
Steinbach G, Lynch PM, Phillips RK et al (2000) The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 342:1946–1952
Elder DJ, Paraskeva C (1999) Induced apoptosis in the prevention of colorectal cancer by non-steroidal anti-inflammatory drugs. Apoptosis 4:365–372
Elder DJ, Paraskeva C (1998) COX-2 inhibitors for colorectal cancer. Nat Med 4:392–393
Jendrossek V, Handrick R, Belka C (2003) Celecoxib activates a novel mitochondrial apoptosis signaling pathway. FASEB J 17:1547–1549
Brown M, Bellon M, Nicot C (2007) Emodin and DHA potently increase arsenic trioxide interferon-alpha-induced cell death of HTLV-I-transformed cells by generation of reactive oxygen species and inhibition of Akt and AP-1. Blood 109:1653–1659
Jendrossek V, Handrick R, Belka C (2003) Celecoxib activates a novel mitochondrial apoptosis signaling pathway. FASEB J 17:1547–1549
Kim H, Rafiuddin-Shah M, Tu HC et al (2006) Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol 8:1348–1358
Hsu YT, Youle RJ (1998) Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J Biol Chem 273:10777–10783
Hsu YT, Youle RJ (1997) Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem 272:13829–13834
Toker A, Yoeli-Lerner M (2006) Akt signaling and cancer: surviving but not moving on. Cancer Res 66:3963–3966
Datta SR, Brunet A, Greenberg ME (1999) Cellular survival: a play in three Akts. Genes Dev 13:2905–2927
Jeong SJ, Pise-Masison CA, Radonovich MF, Park HU, Brady JN (2005) Activated AKT regulates NF-kappaB activation, p53 inhibition and cell survival in HTLV-1-transformed cells. Oncogene 24:6719–6728
Liu Y, Wang Y, Yamakuchi M et al (2001) Phosphoinositide-3 kinase-PKB/Akt pathway activation is involved in fibroblast Rat-1 transformation by human T-cell leukemia virus type I tax. Oncogene 20:2514–2526
Tomita M, Kikuchi A, Akiyama T, Tanaka Y, Mori N (2006) Human T-cell leukemia virus type 1 tax dysregulates β-catenin signaling. J Virol 80:10497–10505
Datta A, Sinha-Datta U, Dhillon NK, Buch S, Nicot C (2006) The HTLV-I p30 interferes with TLR4 signaling and modulates the release of pro- and anti-inflammatory cytokines from human macrophages. J .Biol Chem 281:23414–23424
Han J, Goldstein LA, Gastman BR, Rabinovitz A, Rabinowich H (2005) Disruption of Mcl-1.Bim complex in granzyme B-mediated mitochondrial apoptosis. J Biol Chem 280:16383–16392
Willis SN, Chen L, Dewson G et al (2005) Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 19:1294–1305
Kern MA, Haugg AM, Koch AF et al (2006) Cyclooxygenase-2 inhibition induces apoptosis signaling via death receptors and mitochondria in hepatocellular carcinoma. Cancer Res 66:7059–7066
Cuconati A, Mukherjee C, Perez D, White E (2003) DNA damage response and MCL-1 destruction initiate apoptosis in adenovirus-infected cells. Genes Dev 17:2922–2932
Gradilone A, Silvestri I, Scarpa S et al (2007) Failure of apoptosis and activation on NFkappaB by celecoxib and aspirin in lung cancer cell lines. Oncol Rep 17:823–828
Pang RP, Zhou JG, Zeng ZR et al (2007) Celecoxib induces apoptosis in COX-2 deficient human gastric cancer cells through Akt/GSK3β/NAG-1 pathway. Cancer Lett 251:268–277
Acknowledgment
This work was supported by the National Cancer Institute grant CA106258 to C.N.
Author information
Authors and Affiliations
Corresponding author
Additional information
U. Sinha-Datta and J. M. Taylor have contributed equally to this work.
Rights and permissions
About this article
Cite this article
Sinha-Datta, U., Taylor, J.M., Brown, M. et al. Celecoxib disrupts the canonical apoptotic network in HTLV-I cells through activation of Bax and inhibition of PKB/Akt. Apoptosis 13, 33–40 (2008). https://doi.org/10.1007/s10495-007-0148-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-007-0148-7