
Abstract
Apoptosis is a contributing cause of dopaminergic neuron loss in Parkinson disease. Recent work has shown that erythropoietin (EPO) offers protection against apoptosis in a wide variety of tissues. We demonstrate that exposure of PC12 cells to 1-methyl-4-phenylpyridinium ion (MPP+) with recombinant human EPO, significantly decreased apoptosis as measured by TUNEL and caspase-3 activity when compared to MPP+ treatment alone. EPO induced sustained phosphorylation of Akt and its substrate, GSK-3β, reduced caspase-3 activities in PC12 cells. The anti-apoptotic effect of EPO was abrogated by co-treatment with LY294002, the specific blocker of phosphatidylinositol 3-kinase (PI3K). The effects of EPO on GSK-3β and caspase-3 activities were also blocked by LY294002. LiCl, the inhibitor of GSK-3β, downregulated the caspase-3 activity and blocked the apoptosis induced by MPP+. Finally, we determined that EPO transiently activated the ERK signaling pathway, but PD98059, a specific inhibitor of ERK, does not alter the survival effect of EPO in this model system. Thus, these findings indicate that EPO protects against apoptosis in PC12 cells exposed to MPP+, through the Akt/GSK-3β/caspase-3 signaling pathway, but the ERK pathway is not involved in the EPO-dependent survival enhancing effect in this model system.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Research during the past years has clearly demonstrated that EPO is a potent promoter of neuronal survival. In vitro studies provided most of the information related to the molecular pathways involved in EPO action. These data showed that EPO might have a direct protective role against a variety of neurotoxic insults, such as hypoxic conditions [1], glutamate toxicity [2], free-radical injury [3], and exposure to neurotoxicants [4]. In addition, EPO receptor is abundantly expressed on adult dopaminergic neurons [5], suggesting a direct effect of EPO on neurons. EPO also can protect dopaminergic neurons against MPTP and 6-hydroxydopamine neurotoxicity in vivo and significantly reverse behavioral deficits in mice [6, 7]. Protection by EPO in CNS is mediated through a series of cellular pathways that maintain intricate links between one another.
In humans and nonhuman primates, the neurotoxin 1-methyl-1, 2, 3, 6 tetrahydropyridine (MPTP)-mediated selective damage to dopaminergic neurons of the nigrostriatal pathway has been widely used as a model of Parkinson’s disease [8, 9]. The 1-methyl-4-phenylpyridinium ion (MPP+), the active metabolite of MPTP, stimulates the production of the superoxide radical in vitro and induces cell death in PC12 cells [10]. PC12 cells, a clonal rat pheochromocytoma cell line possess dopamine synthesis, metabolism and transporting systems [11–13], and therefore have been used extensively as a model for studies of MPP+ neurotoxicity and Parkinson’s disease [14–17]. A lot of evidences have demonstrated that MPP+ elicits apoptosis in PC12 cells by activation of caspases [18].
However, up to date, the mechanisms that underlie the protection effect of EPO on the neurotoxicity of MPP+ have not fully been understood. Here, we report that EPO is an effective neuroprotective therapy that promotes survival the PC12 cells in a model of PD in vitro, with reduced apoptotic cell death and inhibition of caspase activation. Moreover, we analyzed in detail the intracellular signal transduction cascades involved in EPO-dependent neuroprotection in this model. Our findings show that the Akt/GSK-3β/caspase-3 dependent pathway plays an important role in mediating the protection of EPO against PC12 cells apoptosis induced by MPP+.
Materials and methods
Reagents
Recombinant Human Erythropoietin Injection was obtained from Shenyang Sunshine Pharmaceutical Co. Ltd. 1-methyl-4-phenylpyridinium (MPP+), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). Kinase inhibitor LY294002 and Lithium chloride (LiCl) were purchased from Sigma-Aldrich, Inc. (St, Louis, MO, USA). Kinase inhibitor PD98059 was obtained from Promega (Mannheim, Germany). The in situ cell death detection kit was from the Boehringer Mannheim, Co. (Mannheim, Germany). BCA kit and Enhanced chemiluminescence (ECL) were purchased from Pierce Chemical Company (Rockford, IL, USA). Phospho-Akt, and Akt antibodies were purchased from Cell Signal Technology, Inc. (Beverly, MA, USA), phospho-ERK and ERK antibodies were obtained from Upstate Biotechnology (NY, USA), phospho-GSK-3β and GSK-3β antibodies were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The Caspase-3 Fluorescent Assay kit was from R&D systems (Minneapolis, MN). Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Gibco BRL (Gaithersburg, MD, USA).
Cell culture
Rat PC12 cells (adrenal gland; pheochromocytoma) were obtained from Chinese Type Culture Collection. PC12 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated calf serum, 5% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin in a water-saturated atmosphere of 5% CO2 at 37°C. The culture medium was changed every 3 days and cells were subcultured about once a week. The medium was changed to serum-deprived medium or medium supplemented with 1% FBS 24 h before experiments and replanted in the 96 and 6 well plates. The next day MPP+ and/or EPO were added to naive PC12 cells. Kinase inhibitors were added to naive PC12 cells preconditioning 1 h if necessary.
Measurement of cell viability
Cell viability was measured by using the MTT assay, which is based on the conversion of MTT to formazan crystals by mitochondrial dehydrogenases [19]. PC12 cells were seeded in 96-well plates at a density of 4 × 104 cells per well and were treated with various concentrations of EPO (0.1, 0.3,1, 3 or 10 U/ml) and 500 μM MPP+ at 37°C. After incubation for up to 24 h, the medium was incubated with 10 μl of 5 mg/ml MTT solution for 4 h. Then the culture medium with MTT was removed and 200 μl DMSO was added to each well to dissolve the formazan. Absorbance was measured at 570 nm (540 nm as a reference) with a model 550-microplate reader. Cell viability was expressed as a percentage of the value in control cultures.
TUNEL assay for apoptotic DNA fragmentation
The DNA fragmentation of apoptotic cells were identified by the TUNEL method which permits the specific labeling of the 3′-OH end of DNA breaks with modified nucleotides by TdT [20]. Briefly, PC12 cells were seeded at a density of 1 × 105 cells/cm2 on coverslips coated with poly-l-lysine and were treated with EPO (1 U/ml) and/or MPP+ (500 μM) at 37°C. After incubation for up to 24 h, the cultures were washed with PBS and fixed with 4% paraformaldehyde in PBS (pH 7.4) for 30 min at room temperature. Endogenous peroxidase was quenched by incubation with 0.3% (v/v) hydrogen peroxide in methanol for 30 min at room temperature and the cells further permeabilized with 0.1% Triton X-100 in 0.1% sodium acetate for 5 min at 4°C. Thereafter, the cells were labeled by incubation with the TUNEL reaction mixture for 60 min at 37°C followed by labeling with peroxidase-conjugated anti-fluorescein anti-goat antibody (Fab fragment) for an additional 30 min. Subsequently, cells were incubated with diaminobenzidine substrate (DAB) to produce a dark brown precipitate and slides were counterstained with hematein stain. For each experiment, all treatments were performed in triplicate wells. Analyzed under light microscopy, the positive staining was identified under the light microscope as dark brown granules. Apoptotic cell death was expressed as “apoptotic index” (number of positively stained apoptotic cells/total number of cells counted × 100%).
Western blot analysis
After exposure to EPO (1 U/ml) and /or MPP+ (500 μM) for 12 h, cells were rinsed twice with cold PBS and lysed in buffer (50 mmol/l Tris–HCl, pH 8.0, 100 mmol/l NaCl, 1 mmol/l EDTA, 1 mmol/l dithiothreitol, 1%Triton X-100, 0.1% SDS, 50 mmol/l sodium fluoride and 1 mmol/l sodium vanadate) containing a protease inhibitor cocktail to obtain whole cell protein. Lysates were cleared by centrifugation and protein concentration was determined by BCA kit. Equal amounts of proteins were fractionated by SDS-polyacrylamide gel electrophoresis, and transferred onto a nitrocellulose membrane. The membranes were blocked with 5% defatted milk in TBS-Tween (TBS-T) (50 mM Tris, pH 7.6, 150 mM NaCl, 0.1% Tween-20) and incubated with anti-phosphospecific Akt, anti-Akt, anti-phosphospecific GSK-3β, anti-GSK-3β, anti-phosphospecific ERK or anti-ERK antibodies (all at 1:1,000 dilution) overnight at 4°C. The signals were detected using goat anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibody and enhanced chemiluminescence (ECL), then exposed to X-ray films (Fuji, Japan).
Measurement of caspase-3 activity
The activity of caspase-3 was detected with the caspase-3 fluorometric assay kit (R&D systems, Minneapolis, MN) according to the instruction manual. This kit uses synthetic tetrapeptide DEVD-AFC as the substrate. In the presence of active caspase-3, the substrate is cleaved between DEVD and AFC, releasing the fluorogenic AFC, which is then detected by spectrofluorometry. The fluorogenic AFC reflects the activity of caspase-3. PC12 cells were incubated in medium containing serum-free medium with MPP+ and/or EPO. Caspase activities were measured in PC12 cells at the indicated time points after MPP+ and/or EPO treatment. After the incubation, cells were collected and lysed in a lysis buffer on ice for 10 min. The protein concentrations of the supernatant fluids were ascertained with the BCA kit. Samples containing 200 μg protein were mixed with the reaction buffer and DEVD-AFC substrate, followed by a 2 h incubation at 37°C. The fluorescence was measured at an excitation wavelength of 400 nm and emission wavelength of 505 nm with fluorometric reader. A caspase-3-like activity was expressed as relative content against that in the cells incubated in the medium containing serum without MPP+.
Statistical analysis
Data are expressed as the mean ± SD. Statistical analyses were performed using one-way analysis of variance (ANOVA) followed by Duncan’s test for multiple comparisons. A probability of P < 0.05 was considered to be statistically significant.
Results
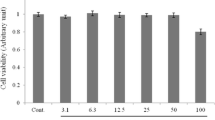
EPO prevents MPP+-induced cell viability loss in PC12 cells in a dose-dependent manner
Our data demonstrated that EPO can promote PC12 cell proliferation even when the cells were cultured without FBS (Fig. 1A) or in the presence of small amounts of FBS (Fig. 1B) in the medium.

Proliferative effect of EPO on cultured PC12 cells. (A) Cells were treated with EPO for 24 h in the medium deprived serum. (B) Cells were treated with EPO for 24 h in the medium supplemented with 1% FBS. Cell viability was assessed by MTT method as described in “Materials and methods”. Data are expressed as percent of values in untreated control cultures, and are means ± SD of three replicate values in four separate experiments. *P < 0.05 vs. controls
To investigate the protective effects of EPO on the cell death induced by MPP+, we tested the neurotoxicity of MPP+ in PC12 cells and cell viability was assessed by the MTT reduction assay. Compared with vehicle controls, exposure to 500 μM MPP+ for 24 h resulted in the loss of 52.5% of PC12 cells.
The cytotoxic effects of 500 μM MPP+ on PC12 cells were significantly blocked by EPO, when the cells were co-incubated with different concentrations (0, 0.1, 0.3,1,3, or 10 U/ml) of EPO and MPP+ for 24 h, Compared with the control group, the survival rate of PC12 cells was 76.8 ± 4.7%, 98.7 ± 9.3%, 112.5 ± 12.5%, 92.9 ± 7.0% and 64.6 ± 5.1%, respectively, when the cells were treated with the indicated concentrations of EPO for 24 h (Fig. 2). The maximal rescue occurred at a concentration of 1 U/ml EPO.

EPO rescued the PC12 cells from MPP+ neurotoxicity. Cell viability was assessed by the MTT method as described in “Materials and methods”. Cells were treated with 500 μM MPP+ for 24 h in the absence or presence of EPO. Data are expressed as percent of values in untreated control cultures, and are means±SD of three replicate values in four separate experiments. ## P < 0.01 vs. controls; *P < 0.05, **P < 0.01 vs. MPP+
EPO application reduces the apoptosis in PC12 cells
In accordance with the previous results [21], the predominant cell death in the MPP+-treated PC12 cells model was apoptosis. To substantiate the anti-apoptotic effect of EPO further, PC12 cells were exposed to MPP+ for 24 h and apoptosis was assayed by TUNEL and caspase activity. MPP+ increased genomic DNA fragmentation and co-incubation of EPO (1 U/ml) decreased the amount of DNA fragmentation as measured by TUNEL (Fig. 3A). Caspase 3-like activity was significantly increased in PC12 cells exposed in MPP+ compared to controls. EPO (1 U/ml) abrogated the increase in caspase 3-like activity from 240.0 ± 14.6% to 130.0 ± 14.4%, P < 0.05, Fig. 3B).

EPO protected PC12 cells from MPP+-induced apoptosis. PC12 cells were exposed to 500 μM MPP+ for 24 h in the presence of EPO (1 U/ml) and apoptosis was assayed by TUNEL and caspase activity. (A) Treatment with EPO decreased DNA fragmentation significantly during exposure to MPP+ (## P < 0.01 vs. control;**P < 0.01 vs. MPP+, n = 6). (B) EPO prevents MPP+-induced increase in caspase-3 activity (## P < 0.01 vs. control; **P < 0.01 vs. MPP+, n = 6)
The PI3K/Akt pathway was activated in MPP+-treated PC12 cells by EPO and mediated EPO-induced neuroprotection
Akt is a pro-survival kinase and is activated by the phosphorylation at Ser473 via the PI3K pathway, which is thought to be one of the molecules that promotes cell survival and prevents apoptosis [22, 23]. To evaluate a possible mechanism by which EPO prevents cellular death in our model, we measured the changes in phosphorylation levels of Akt in the PC12 cells exposed to EPO in the presence or absence of MPP+.
As shown in Fig. 4, the level of phospho-Akt was low in PC12 cells under unchallenged condition. Enhanced levels of phospho-Akt were detected 30 min after EPO was added to the culture, and levels remained elevated for at least 12 h (Fig. 4A). There was a significant increase in the phosphorylation of Akt in MPP+-treated cells by EPO, when EPO was added to cultures simultaneously with MPP+, it maintained increased phospho-Akt levels for at least 12 h (Fig. 4B). To confirm the involvement of Akt further, we inhibited the upstream pathway that controls Akt activation. The PI3K inhibitor, LY294002 (10 μM) was added to PC12 cells 1 h before EPO. LY294002 thoroughly abolished EPO-induced phosphorylation of Akt, indicating that EPO activates Akt likely through PI3-kinase (Fig. 4C). Total Akt levels in each experiment remained constant (Fig. 4, Akt lanes).

EPO induced phosphorylation of Akt in PC12 cells. (A) Representative Western blots showing the protein levels of phosphorylated and total Akt in PC12 cells at serial time points. Western blot analysis of PC12 cells stained for activated phospho-Akt (p-Akt) shows that EPO quickly induces a sustained activation of Akt, lasting at least 12 h. (B) When MPP+ was also added to the cell medium, phospho-Akt was elevated at all time points following MPP+ exposure. (C) EPO-induced phosphophorylation of Akt in PC12 cells was suppressed by PI3K inhibitor LY294002 3 h after EPO and/or MPP+ exposure. The blots presented are representatives of three independent experiments with similar results. In all blots, staining for total Akt was used as a loading control
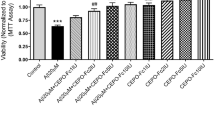
This effect of rescuing the cell from death induced by EPO was lost with addition of LY 294002 (10 μM), a specific inhibitor of PI3K (Fig. 5). LY294002 also abrogated the anti-apoptotic effect of EPO (Fig. 10) as measured by DNA fragmentation on TUNEL analysis. The result indicated that Akt is activated by EPO regardless of MPP+ and plays an important role in EPO-mediated neuroprotection.

PI3K inhibitor blocked the protective effect of EPO on MPP+-induced neurotoxicity. PC12 cell viability was measured using the MTT assay. Pre-incubation with the specific PI-3 kinase inhibitor LY 294002 (LY, 10 μM) resulted in the complete elimination of the EPO (1 U/ml)-induced neuroprotection. LY294002 alone did not alter cell viability in control cells. Data are means ± SD of three replicate values in four separate experiments. ## P < 0.01 vs. controls; **P < 0.01 vs. MPP+; ++ p < 0.01 vs. MPP+ plus EPO
The substrate GSK-3β was activated by EPO
The anti-apoptotic function of Akt was induced allowing the phosphorylation of its downstream substrates, GSK-3β [22, 23]. The main regulatory mechanism of these enzymes is by phosphorylation: Akt is activated while GSK-3β is inhibited by phosphorylation [24, 25]. The inactivation of GSK-3β can result in the prevention or reduction in apoptotic injury in neurons [26]. Therefore, we next investigated whether EPO enhances the phosphorylation of GSK-3β in PC12 cells treated by MPP+.
As shown in Fig. 6A, phosphorylation of GSK-3β at Ser9 was enhanced 1 and 6 h following EPO-treated. The time course of enhanced phosphorylation of GSK-3β is in parallel with the activation of Akt (Fig. 4A, 6A).

Epo increased the phosphorylation of GSK-3β in PC12 cells. (A) Representative Western blots showing protein levels of phosphorylated and total GSK-3β in PC12 cells at serial time points as indicated. (B) The average levels of phospho-GSK-3β in PC12 cells determined by Western blot were increased at all time points in the presence of MPP+, compared with the control group. (C) EPO-induced phosphophorylation of GSK-3β in PC12 cells was suppressed by PI3K inhibitor LY294002 3 h after EPO and/or MPP+ exposure. The blots presented are representatives of three independent experiments with similar results. In all blots, staining for total GSK-3β was used as a loading control
Figures 3B and 5B also show concurrent increase in Akt and GSK-3 phosphorylation in MPP+-treated cells by EPO. LY294002 pretreatment abolished phosphorylation of both Akt and GSK-3β by EPO (Figs. 4C, 6C). Furthermore, inhibition of GSK-3β by LiCl (20 mmol/l) promoted viability and reduced apoptosis in PC12 cells subjected to MPP+ (Figs. 7, 10). These data indicate that GSK-3β was the major substrate of Akt contributing to the EPO-mediated neuroprotection against neurotoxicity of MPP+.

GSK-3 inhibitor prevented MPP+-induced neurotoxicity. PC12 cell viability was measured using the MTT assay. Pre-incubation with the specific GSK-3β inhibitor LiCl (20 mM) resulted in the complete elimination of the MPP+ (500 μM)-induced neurotoxicity. LiCl alone did not alter cell viability in control cells. Data are means ± SD of three replicate values in four separate experiments. ## P < 0.01 vs. controls; **P < 0.01 vs. MPP+; ++ P < 0.01 vs. MPP+ plus EPO
The ERK pathway was not activated in MPP+-treated PC12 cells by EPO, and not involved in EPO-induced neuroprotection
The MAPK/ERK pathway plays dual roles in the regulation of cell apoptosis. Downstream kinases activated by MAPK and associated with cell survival are the ERK proteins, whereas those most often linked to cell death are JNK and p-38 [27]. We next investigated whether ERK play a role in the EPO-mediated neuroprotection.
No significant difference was detected in the levels of p-ERK between control and EPO-treated groups. EPO caused the increase in p-ERK1/2 levels, which began within 30 min after MPP+ exposure and subsided to control levels at 6 h (Fig. 8A). The phosphorylation of ERK displayed a transient increase at 12 h after treated with MPP+ in the EPO-treated group compared with the control group (Fig. 8B). The phosphorylation of the kinase was completely inhibited by PD98059. In these experiments, total ERK levels did not change (ERK lanes in Fig. 8).

EPO transiently increased ERK1/2 activity in PC12 cells. (A) Western blot demonstrating transiently enhanced levels of phosphorylated ERK1/2 (p-ERK1/2) in cells induced by Epo (1 U/ml). (B) Western blot shows that the presence of both MPP+ and EPO (1 U/ml) had no additive effect on p-ERK1/2 induction. (C) Western blot showing that EPO-induced increases in p-ERK1/2 were diminished in the presence of the MAPK inhibitor PD98059 (50 μM). The blots presented are representatives of three independent experiments with similar results. In all blots, staining for total ERK1/2 was used as a loading control (ERK1/2)
As EPO enhanced p-ERK1/2 levels, we further investigated the role of this signaling pathway using ERK inhibitors, PD98059 (50 μM). However, PD98059 did not prevent the neuroprotective effect of EPO (Figs. 9, 10). Our results slightly deviated from the previous report [7], which suggested that activation of ERK1/2 contributes moderately to the neuroprotective effect of EPO. These results suggest that the ERK pathway did not involved in EPO-induced neuroprotection.

EPO-mediated neuroprotection was not affected by treatment with the ERK inhibitor PD 98059 (PD). PC12 cell viability was measured using the MTT assay. Pre-incubation with the specific ERK inhibitor PD98059 (PD, 50 μM) did not resulted in the elimination of the EPO (1 U/ml)-induced neuroprotection. PD98059 alone did not alter cell viability in control cells. Data are means ± SD of three replicate values in four separate experiments. ## P < 0.01 vs. controls; **P < 0.01 vs. MPP+

Detection of apoptotic cell death using TUNEL staining. A, control; B, 500 μM MPP+ (M) only; C, 500 μM MPP+ plus 1 U/ml EPO (M + E); D, 1 U/ml EPO only (E); E, 500 μM MPP+ plus 1 U/ml EPO with 10 μM LY294002 (M + E + LY); F, 500 μM MPP+ plus 1 U/ml EPO with 50 μM PD98059 (M + E + PD); G, 500 μM MPP+ plus 20 mM LiCl (M + LiCl). EPO and LiCl significantly reduced the number of TUNEL positive cells (dark brown staining) relative to MPP+ (M). In application of LY294002 into the cells eliminated the protection of EPO. Values are mean ± SD for six individual experiments. ## P < 0.01 vs. controls; **P < 0.01 vs. MPP+; ++ P < 0.01 vs. MPP+ plus EPO
Akt/GSK-3β pathway prevented apoptosis by blocking activity of caspase-3 in MPP+-treated cells
The mechanism of the progression of DNA fragmentation was clarified, and it was found that caspase activity was necessary. The increasing findings have shown that the activation of caspase-3 is postulated to contribute to dopaminergic cell death in the MPP+-induced neurodegeneration [28]. As shown in Fig. 11, we also found that MPP+ induced a time dependent increase in caspase-3-like proteinase activities (Fig. 11). EPO significantly decreased caspase 3-like activities induced by MPP+ toxicity. To estimate further the contribution of the Akt pathway to the caspase activity that induces DNA fragmentation, caspase-3-like protease activity was measured with or without LY294002. Consistent with the critical role of PI3K in mediating EPO -induced neuroprotection, the addition of the PI3K inhibitor, LY 294002 (10 μM), in PC12 cells reversed the effect of EPO on caspase-3 activation (Fig. 11). In contrast, LiCl, the inhibitor of GSK-3β, downregulated the caspase-3 activity induced by MPP+. These results indicate a downstream event of caspase-3 activation after Akt-GSK signaling.

Caspase-3-like activity was involved in MPP+-induced cell death, and EPO suppressed caspase-3-like activity. Cells were maintained as described in Fig. 1, and the medium was changed to serum-free DMEM containing 500 μM MPP+ (M) or with 1 U/ml EPO (M+E) or with 1 U/ml EPO plus 10 μM LY294002 (M + E + LY) or with 20 mmol/l LiCl (M + LiCl). After the addition of reagents, the intracellular caspase-3-like protease activity was measured as described in “Materials and methods”. Cells exposed to MPP+ alone showed a time-dependent increase in caspase-3-like activities. Treatment with EPO blocked MPP+-induced activation of caspase-3 and the GSK-3 inhibitor, LiCl significantly diminished the activities at all time points. The caspase inhibitory effect of EPO was reversed in the presence of the PI3K inhibitor, LY 294002. Values are means±SD for six individual experiments, and statistical analysis was carried out at each time point by ANOVA. **P < 0.01 vs. MPP+; ## P < 0.01 vs. MPP+ plus EPO. These data represent three independent experiments
These results suggested that the Akt pathway suppressed caspase-3-like activity in the late phase of MPP+-induced apoptosis and that Akt helped to prevent cell death. EPO increase GSK-3β phosphorylation and thus inactivation of GSK-3β activity, leading to inhibition of caspase-3 activation.
Discussion
Excessive apoptosis is thought to be one of the key factors contributing to Parkinson’s disease. Neuronal cell death due to 1-methyl-4-phenylpyridinium (MPP+) is mediated by opening of the mitochondrial permeability transition pore, release of cytochrome c and activation of caspases [29]. Once released to the cytosol, cytochrome c could form apoptosome together with apoptosis-activating factor Apaf-1 and procaspase-9, leading to the activation of capase-9, and then activate capase-3 [30]. Caspase-3 has been demonstrated to participate in MPP+-induced apoptosis [31]. Moreover, some previous works showed that reactive oxygen species (ROS) is also implicated in MPP+-induced cytotoxicity including mitochondrial permeability transition pore opening and cytochrome c release [32, 33].
In the present study, we demonstrated that EPO prevented the degeneration of PC12 cells induced by MPP+. As others have found in various cell culture models of neuronal cell death [34, 35], the survival-promoting action of EPO followed a bell-shaped dose–response curve in our in vitro paradigm. Increasing EPO concentration in the cell culture medium above an optimal dose resulted in a decline of the cell rescue rate. However, we did not observe further protective effect at the higher concentration. Several studies were then able to confirm this particular dose–response behavior of EPO in vivo [36, 37], which might have relevance for the design of upcoming clinical trials. EPO’s effects in clinical studies, as in the recent human stroke study that demonstrated beneficial effects in the context of cerebral ischemia [38], may be further improved by an optimized dose regimen in the future.
EPO has effects through binding EPO receptor which has been detected in PC12 cells [39]. Once it is bound to its receptor, EPO initiates the downstream signaling pathways. In the present study, we showed that EPO protects against PC12 cells apoptosis through activation of the Akt/GSK-3β/caspase-3 mediated signaling mechanism. Treatment of recombinant EPO in PC12 cells activates Akt, leading to GSK-3β phosphorylation. Inactivation of GSK-3β by Akt-mediated phosphorylation led to inhibition of caspase-3 activation and thus attenuation of apoptosis. These findings provide new insights into the role and signaling mechanisms mediated by EPO in protection against MPP+-induced PC12 cells injury and apoptosis and may have significance in the development of molecular targets for future therapeutic application.
PI3K/Akt is a well-known signaling pathway involved in cell protection under various stresses, including oxidative stress. PI3K/Akt pathway promotes cardiomyocytes survival against oxidative stress-induced apoptosis [40] and also delivers an anti-apoptotic signal in PC12-ErbB4 cells against oxidative stress induced by MPP+ [41]. In an attempt to study the molecular mechanism that could be involved in the neuroprotection of EPO, we investigated the activation of Akt, the direct effector downstream of PI3-K in this signal transduction cascade [42]. EPO induced the high and sustained level of activation of Akt in PC12 cells. The increase in p-Akt mediated by EPO was dependent on unimpaired by EPO was dependent on unimpaired functioning of the upstream pathway involving PI3 kinase. The PI3K inhibitor, LY294002, abolished the increase in p-Akt and the anti-apoptotic effect of EPO. Our results identified the PI3K/Akt signaling pathway as the one responsible for protecting PC12 cells from apoptosis.
Following the PI3-K/Akt pathway, we also examined Glycogen synthase kinase-3β (GSK-3β), one downstream target of Akt [43]. GSK-3β regulates a variety of elements involved in cellular metabolism, including glycogen synthase [43, 44]. It is a constitutively active kinase in cells and is primarily regulated through inhibition of its activity. Akt can phosphorylate GSK-3β and inactivate the enzyme [45], which can occur during neuronal degeneration [46]. GSK-3β has also been shown to regulate cell survival in PC12 cells: transfection of constitutively active GSK-3 drives PC12 cells into apoptosis, whereas transfection with kinase-inactive GSK-3 blocks apoptosis [47].
Using MPP+-induced PC12 cells death model, we presented convincing evidence that treatment of EPO activates Akt, resulting in the inhibition of GSK-3 activity in the anti-apoptotic process. In agreement with previous report showing that GSK-3β activity is suppressed by EPO in erythroid progenitors [48], our data demonstrated EPO was able to increase the level of the inactive form of GSK-3β in parallel with the activation of Akt. We also showed that PI3K inhibitor, LY294002, blocked EPO-induced phosphorylation of GSK-3β, and inhibition of GSK-3β by LiCl promoted viability and reduced apoptosis in PC12 cells subjected to MPP+. These results indicate that the protective effect of EPO on apoptosis is also mediated by PI3K/Akt-dependent GSK-3β phosphorylation. Thus, both activation of Akt and inactivation of GSK-3 by EPO are consistent with promoting cell survival in PC12 cells. Phosphorylation of GSK-3β may represent another mechanism by which EPO decreases apoptosis in PC12 cells.
Although the mechanism by which inhibition of GSK-3β through PI3K/Akt suppresses apoptosis is not known. GSK-3β has been shown to participate in apoptosis in several cell types and is known to be an upstream regulator of programmed cell death [49]. GSK-3β has been demonstrated to induce caspase-3 activation and activate the proapoptotic tumor suppressor gene, p53 [50]. It also has been suggested that GSK-3β promotes activation and translocation of the proapoptotic Bcl-2 family member, Bax [48, 51], which, upon aggregation and mitochondrial localization, induces cytochrome c release.
Caspase-3 is thought to be activated during the final step of the proapoptotic signaling pathway in many cell lines, and it represents a potential therapeutic target for suppression of PC12 cells apoptosis induced by MPP+ [52–54]. In dopaminergic cell , MPP+ induced apoptosis is associated with release of cytochrome c and activation of caspase-3 [28], whereas inhibition of caspase activity has been shown to attenuate apoptosis. In the present study, we confirmed that EPO-mediated Akt signaling effects in regulating PC12 cells death is executed by inhibition the caspase-3 activity. Previous studies in human neuroblastoma cell lines showed that the increase activity of GSK-3 preceded activation of the caspase cascade [50]. Consistent with the critical role of Akt/GSK-3β in mediating EPO-induced neuroprotection, our results indicated that the addition of LY 294002 reversed the effect of EPO on caspase-3 activation, whereas LiCl, the inhibitor of GSK-3β, downregulated the caspase-3 activity induced by MPP+. These results indicate activation of caspase-3 is a downstream event after Akt/GSK-3β signaling.
EPO also has the ability to activate the extracellular signal-regulated kinases (ERKs) pathway, which are important for neuron survival [55]. It is not clear whether the survival signal pathway of EPO is restricted by the PI3-K/Akt pathway or co-exists with the ERKs pathway in MPP+-induced apoptosis. PD98059, a specific inhibitor of the activation of ERKs, was used to settle this issue [56, 57]. These pharmacological experiments suggest that EPO active ERK1/2 transiently and the activity of ERKs was decreased by the addition of PD98059. However, PD98059 does not affect the survival effect of EPO in MPP+-induced apoptosis. These results indicate that ERKs is not involved in the survival pathway of EPO in MPP+-induced apoptosis.
With regard to the role of ERK1/2 in the EPO-mediated neuroprotection, the outcomes of recent studies show no consistency. ERK was reported to be unimportant in the mediation of EPO’s neuroprotective effect against cerebral ischemia [58], cardiac ischemia [59] and Parkinson’s disease [7]. In contrast, ERK was considered to be crucial for the neuroprotection of EPO against cerebral ischemia [60–62]. The relative contributions of ERK1/2 and PI3K in EPO-induced neuroprotection are ambiguous, depending on the model system examined. Our results support the notion that EPO-mediated neuroprotection in the MPP+ treatment PC 12 cells occurs primarily via activation of the Akt/GSK-3β/caspase-3 and is not attributable to the activation of the ERK1/2 pathway.
Conclusion
Taken together, these data demonstrate that EPO exerted protection against apoptosis induced by MPP+ in PC12 cells. This work re-emphasizes the importance of Akt in protective signaling, and demonstrates that EPO-induced neuroprotection is mediated by the Akt/GSK-3β signaling pathways. Our results also implicate caspase-3 activation as a critical downstream signaling effector in EPO/PI3K/Akt/GSK-3β signaling. In conclusion, the elucidation of the neuroprotective action of EPO in the current study adds to a growing literature suggesting the potential beneficial role for this factor in Parkinson disease.
References
Marti HH, Wenger RH, Rivas LA et al (1996) Erythropoietin gene expression in human monkey and murine brain. Eur J Neurosci 8:666–676
Morishita E, Masuda S, Nagao M, Tasuda Y, Sasaki R (1997) Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate induced neuronal death. Neuroscience 76:105–116
Chong ZZ, Lin SH, Kang JQ, Maiese K (2003) Erythropoietin prevents early and late neuronal demise through modulation of Akt1 and induction of caspase 1, 3, and 8. J Neurosci Res 71:659–669
Villa P, Bigini P, Mennini T et al (2003) Erythropoietin selectively attenuates cytokine production and inflammation in cerebral ischemia by targeting neuronal apoptosis. J Exp Med 198:971–975
Csete M, Rodriguez L, Wilcox M, Chadalavada S (2004) Erythropoietin receptor is expressed on adult rat dopaminergic neurons and erythropoietin is neurotrophic in cultured dopaminergic neuroblasts. Neurosci Lett 359:124–126
Genc S, Kuralay F, Genc K et al (2001) Erythropoietin exerts neuroprotection in 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-treated C57/BL mice via increasing nitric oxide production. Neurosci Lett 298:139–141
Signore AP, Weng Z, Hastings T et al (2006) Erythropoietin protects against 6-hydroxydopamine-induced dopaminergic cell death. J Neurochem 96:428–443
Heikkila RE, Hess A, Duvoisin RC (1984) Dopaminergic neurotoxicity of 1-methyl-4-phenyl-1, 2, 5, 6-tetrahydropyridine in mice. Science 224:1451–1453
Langston JW, Ballard P, Tetrud JW, Irwin I (1983) Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219:979–980
Itano Y, Kitamura Y, Nomura Y (1994) 1-Methyl-4-phenylpyridinium (MPP+)-induced cell death in PC12 cells. Neurochem Int 25:419–424
Rebois RV, Reynolds EE, Toll L, Howard BD (1980) Storage of dopamine and acetylcholine in granules of PC12, a clonal pheochromocytoma cell line. Biochemistry 19:1240–1248
Hatanaka H (1981) Nerve growth factor-mediated stimulation of tyrosine hydroxylase activity in a clonal rat pheochromocytoma cell line. Brain Res 222:225–233
Tuler SM, Hazen AA, Bowen JM (1989) Release and metabolism of dopamine in a clonal line of pheochromocytoma (PC12) cells exposed to fenthion. Fund Appl Toxicol 13:484–492
Mutoh T, Tokuda A, Marini AM, Fujiki N (1994) 1-Methyl-4-phenylpyridinum kills differentiated PC12 cells with a concomitant change in protein phosphorylation. Brain Res 661:51–55
Cappelletti G, Maggioni MG, Maci R (1999) Influence of MPP+ on the state of tubulin polymerisation in NGF-differentiated PC12 cells. J Neurosci Res 56:28–35
Gelinas S, Martinoli MG (2002) Neuroprotective effect of estradiol and phytoestrogens on MPP+-induced cytotoxicity in neuronal PC12 cells. J Neurosci Res 70:90–96
Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA (2002) Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease. J Neurosci 22:10690–10698
Viswanath V, Wu Y, Boonplueang R et al (2001) Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease. J Neurosci 21:9519–9528
Yamamoto T, Yuyama K, Nakamura K, Kato T, Yamamoto H (2000) Kinetic characterization of the nitric oxide toxicity for PC12 cells: effect of half-life time of NO release. Eur J Pharmacol 397:25–33
Ohgoh M, Kimura M, Ogura H, Katayama K, Nishizawa Y (1998) Apoptotic cell death of cultured cerebral cortical neurons induced by withdrawal of astroglial trophic support. Exp Neurol 149:51–63
Hartley A, Stone JM, Heron C, Cooper JM, Schapira AH (1994) Complex I inhibitors induce dose-dependent apoptosis in PC12 cells: relevance to Parkinson’s disease. J Neurochem 63:1987–1990
Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C (2003) PI3K/Akt and apoptosis: size matters. Oncogene 22:8983–8998
Amaravadi R, Thompson CB (2005) The survival kinases Akt and Pim as potential pharmacological targets. J Clin Invest 115:2618–2624
Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heidereich KA (2000) Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3beta. Mol Cell Biol 20:9356–9363
Kaytor MD, Orr HT (2002) The GSK3 beta signaling cascade and neurodegenerative disease. Curr Opin Neurobiol 12:275–278
Alvarez G, Munoz-Montano JR, Satrustegui J, Avila J, Bogonez E, Diaz-Nido J (1999) Lithium protects cultured neurons against betaamyloid-induced neurodegeneration. FEBS Lett 453:260–264
Liou AK, Clark RS, Henshall DC, Yin XM, Chen J (2003) To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: a review on the stress-activated signaling pathways and apoptotic pathways. Prog Neurobiol 69:103–142
Han BS, Hong HS, Choi WS, Markelonis GJ, Oh TH, Oh YJ (2003) Caspase-dependent and -independent cell death pathways in primary cultures of mesencephalic dopaminergic neurons after neurotoxin treatment. J Neurosci 23:5069–5078
Seaton TA, Cooper JM, Schapira AH (1997) Free radical scavengers protect dopaminergic cell lines from apoptosis induced by complex I inhibitors. Brain Res 777:110–118
Thornberry NA, Lazebnik Y (1998) Caspases: enemies within. Science 281:1312–1316
Blum D, Torch S, Lambeng N et al (2001) Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson’s disease. Prog Neurobiol 65:135–172
Di Monte D, Sandy MS, Ekstrom G, Smith MT (1986) Comparative studies on the mechanisms of paraquat and 1-methyl-4-phenylpyridine (MPP+) cytotoxicity. Biochem Biophys Res Commun 137:303–309
Cassarino DS, Parks JK, Parker WD Jr, Bennett JP Jr (1999) The parkinsonian neurotoxin MPP+ opens the mitochondrial permeability transition pore and releases cytochrome c in isolated mitochondria via an oxidative mechanism. Biochem Biophys Acta 1453:49–62
Siren AL, Fratelli M, Brines M et al (2001) Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci USA 98:4044–4049
Chong ZZ, Lin SH, Kang JQ, Maiese K (2003) Erythropoietin prevents early and late neuronal demise through modulation of Akt1 and induction of caspase 1, 3, and 8. J Neurosci Res 71:659–669
Sakanaka M, Wen TC, Matsuda S et al (1998) In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci USA 95:4635–4640
Weishaupt JH, Rohde G, Polking E, Siren AL, Ehrenreich H, Bahr M (2004) Effect of erythropoietin axotomy-induced apoptosis in rat retinal ganglion cells. Invest Ophthalmol Vis Sci 45(5):1514–1522
Ehrenreich H, Hasselblatt M, Dembowski C et al (2002) Erythropoietin therapy for acute stroke is both safe and beneficial. Mol Med 8:495–505
Masuda S, Nagao M, Takahata K et al (1993) Functional erythropoietin receptor of the cells with neural characteristics. Comparison with receptor properties of erythroid cells. J Biol Chem 268:11208–11216
Hong F, Kwon SJ, Jhun BS et al (2001) Insulin-like growth factor-1 protects H9c2 cardiac myoblasts from oxidative stress-induced apoptosis via phosphatidylinositol 3-kinase and extracellular signal-regulated kinase pathways. Life Sci 68:1095–1105
Di Segni A, Farin K, Pinkas Kramarski R (2006) ErbB4 activation inhibits MPP+-induced cell death in PC12-ErbB4 cells: involvement of PI3K and Erk signaling. J Mol Neurosci 29:257–268
Songyang Z, Baltimore D, Cantley LC, Kaplan DR, Franke TF (1997) Interleukin-3-dependent survival by the Akt protein kinase. Proc Natl Acad Sci USA 94:11345–11350
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378:785–789
Hajduch E, Alessi DR, Hemmings BA, Hundal HS (1998) Constitutive activation of protein kinase B alpha by membrane targeting promotes glucose and system A amino acid transport, protein synthesis, and inactivation of glycogen synthase kinase 3 in L6 muscle cells. Diabetes 47:1006–1013
Shaw M, Cohen P, Alessi DR (1997) Further evidence that the inhibition of glycogen synthase kinase-3beta by IGF-1 is mediated by PDK1/PKBinduced phosphorylation of Ser-9 and not by dephosphorylation of Tyr-216. FEBS Lett 416:307–311
Bhat RV, Shanley J, Correll MP et al (2000) Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3-beta in cellular and animal models of neuronal degeneration. Proc Natl Acad Sci 97:11074–11079
Pap M, Cooper GM (1998) Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem 273:19929–19932
Somervaille TC, Linch DC, Khwaja A (2001) Growth factor withdrawal from primary human erythroid progenitors induces apoptosis through a pathway involving glycogen synthase kinase-3 and Bax. Blood 98:1374–1381
Bijur GN, Jope RS (2001) Proapoptotic stimuli induce nuclear accumulation of glycogen synthase kinase-3 beta. J Biol Chem 276:37436–37442
King TD, Bijur GN, Jope RS (2001) Caspase-3 activation induced by inhibition of mitochondrial complex I is facilitated by glycogen synthase kinase-3beta and attenuated by lithium. Brain Res 919:106–114
Yamaguchi H, Wang HG (2001) The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene 20:7779–7786
Bo J, Ming BY, Gang LZ, Lei C, Jia AL (2005) Protection by puerarin against MPP+-induced neurotoxicity in PC12 cells mediated by inhibiting mitochondrial dysfunction and caspase-3-like activation. Neurosci Res 53:183–188
Lee CS, Han ES, Kim YK (2006) Piperine inhibition of 1-methyl-4-phenylpyridinium-induced mitochondrial dysfunction and cell death in PC12 cells. Eur J Pharmacol 537:37–44
Guan S, Jiang B, Bao YM, An LJ (2006) Protocatechuic acid suppresses MPP (+)-induced mitochondrial dysfunction and apoptotic cell death in PC12 cells. Food Chem Toxicol 44:1659–1666
Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME (1995) Opposing effects of Erk and Jnk-P38 Map kinases on apoptosis. Science 270:1326–1331
Virdee K, Tolkovsky AM (1996) Inhibition of p42 and p44 mitogenactivated protein kinase activity by PD98059 does not suppress nerve growth factor-induced survival of sympathetic neurons. J Neurochem 67:1801–1805
Yan CYI, Greene LA (1998) Prevention of PC12 cell death by N-acetylcysteine requires activation of the Ras pathway. J Neurosci 18:4042–4049
Ruscher K, Freyer D, Karsch M et al (2002) Erythropoietin is a paracrine mediator of is ischemic tolerance in the brain: evidence from an in vitro model. J Neurosci 22:10291–10301
Hanlon PR, Fu P, Wright GL, Steenbergen C, Arcasoy MO, Murphy E (2005) Mechanisms of erythropoietin-mediated cardioprotection during ischemia-reperfusion injury: role of protein kinase C and phosphatidylinositol 3-kinase signaling. FASEB J 19:1323–1325
Siren AL, Fratelli M, Brines M et al (2001) Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci 98:4044–4049
Lee SM, Nguyen TH, Park MH et al (2004) EPO receptor-mediated ERK kinase and NF-kappaB activation in erythropoietin-promoted differentiation of astrocytes. Biochem Biophys Res Commun 320:1087–1095
Kilic E, Kilic U, Soliz J, Bassetti CL, Gassmann M, Hermann DM (2005) Brain-derived erythropoietin protects from focal cerebral ischemia by dual activation of ERK-1/-2 and Akt pathways. FASEB J 19:2026–2028
Acknowledgements
The authors gratefully acknowledge Jieping Zhou for technical assistance. This study was supported by a grant from the Program of National Natural Science Foundation of China (No. 30570627/C030307).
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors Yan Wu and You Shang are equally contributed to this work.
Rights and permissions
About this article
Cite this article
Wu, Y., Shang, Y., Sun, S. et al. Erythropoietin prevents PC12 cells from 1-methyl-4-phenylpyridinium ion-induced apoptosis via the Akt/GSK-3β/caspase-3 mediated signaling pathway. Apoptosis 12, 1365–1375 (2007). https://doi.org/10.1007/s10495-007-0065-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-007-0065-9