
Abstract
Labd-14-ene-8, 13-diol (sclareol) is a labdane-type diterpene, which has demonstrated significant cytotoxic activity against human leukemic cell lines, but its effect on solid tumor-derived cells is uknown. Here, we demonstrate that addition of sclareol to cultures of human colon cancer HCT116 cells results in inhibition of DNA synthesis, arrest of cells at the G1 phase of the cell cycle, activation of caspases-8, -9, PARP degradation, and DNA fragmentation, events characteristic of induction of apoptosis. Intraperitoneal (ip) administration of sclareol alone, at the maximum tolerated dose, was unable to induce suppression of growth of HCT116 tumors established as xenografts in immunodeficient SCID mice. In contrast, ip administration of liposome-encapsulated sclareol, following a specific schedule, induced suppression of tumor growth by arresting tumor cell proliferation as assessed by detecting the presence of the cell proliferation-associated nuclear protein, Ki67, in thin tumor sections. These findings suggest that sclareol incorporated into liposomes may possess chemotherapeutic potential for the treatment of colorectal and other types of human cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A large number of the phytochemicals, diterpenoids, abundantly occur in nature [1]. Labdane diterpenes abundant in several families of plants and algae possess a labdane skeleton consisting of a decalin system and a C-6 ring, which may be open or closed with an oxygen atom, as in manoyl oxide and its derivatives [2, 3]. Of special interest among these compounds that exhibit a variety of activities [4] is forskolin, a manoyl oxide derivative that can activate adenylate cyclase by directly interacting with the catalytic subunit or a closely associated protein of the adenylate cyclase system [5]. Because of its unique biochemical properties, forskolin is an invaluable tool for investigations of the role of c-AMP in biological processes. Further, several labdane diterpenes are cytotoxic and cytostatic for human leukemia [6–11], breast [12], and colon [13] cancer cell lines.
The ditertiary alcohol, labd-14-ene-8, 13-diol (sclareol), which was first isolated from the plant Salvia sclarea [14], is abundant in nature, and is used as a fragrance in cosmetics, flavoring additive in food and beverage industry, and folk medicine. Sclareol induces apoptosis in human leukemia cell lines, via an interesting mechanism that down-regulates c-myc without affecting the expression of the anti-apoptotic protein, Bcl-2 [9], and in cells derived from solid tumors by a mechanism that appears to be independent on p53-expression [12]. Further, in a preliminary study we have shown that sclareol possesses a modest anticancer activity in a human colon cancer xenograft/mouse model, when this compound is administered in an encapsulated (liposomal) form, as this was concluded calculating the doubling time of tumors from 200 to 400 mm3 [13]. Our choice of liposomes as lipid-based drug carriers to deliver sclareol was motivated by their well documented ability to deliver bioactive compounds in a targeted manner, and their favourable physicochemical properties that typically result in improvement of the pharmacokinetic properties and pharmacological response of the encapsulated drugs, while reducing drug-associated side effects (for recent reviews, see refs. [20–24]).
In most cases, anticancer drug treatment results in the activation of caspases, enzymes which effectively execute various forms of cell death by apoptosis. The death receptor-dependent apoptotic pathway is triggered at the cell surface and requires activation of caspase-8, whereas the mitochondrion-dependent pathway is initiated with release of mitochondrial cytochrome c into the cytoplasm and requires activation of caspase-9. Subsequently, caspase-8 or -9 can activate caspase-3, which in turn targets and degrades specific and vital cellular proteins, ultimately resulting in nuclear DNA degradation and apoptotic death of the cells (for reviews on caspases, see refs. [15–17]). However, it is not unusual for the activated pathway to display only some of the characteristics of the “classical” apoptotic pathways [18, 19].
Our initial published observations and results [13] prompted us to investigate more extensively the effect of sclareol on the cell cycle of human colon cancer cells in vitro. Furthermore, and since the apoptotic pathway activated by sclareol was still unknown, we sought to assess whether sclareol's death inducing activity is mediated via an established apoptosis pathway. Finally, we wanted to investigate conditions that could induce significant suppression or regression of HCT116 tumors.
In this report, we describe experimental findings to demonstrate for the first time that (i) sclareol can induce G1-phase arrest and apoptosis in HCT116 colon cancer cells in vitro associated with activation of caspases-8, -9 and -3, and, (ii) sclareol alone at the maximum tolerated dose (MTD) is inactive, whereas liposome-encapsulated sclareol, at doses much higher than the MTD of sclareol alone, can suppress colon tumor growth presumably by inhibiting tumor cell proliferation.
Materials and methods
Chemicals/biological reagents
Trichloroacetic acid (TCA), sulphorhodamine B (SRB), Trizma base, acetic acid, sclareol, rabbit polyclonal antibodies to caspase-8 (GD-13), caspase-3 (CPP32), mouse monoclonal antibody to PARP (clone C-2-10), the secondary antibodies, peroxidase-conjugated anti-mouse and anti-rabbit IgG, and the chromogen, 3,3′-diaminobenzidine (DAB), were purchased from Sigma-Aldrich (St. Louis, MO, USA). Rabbit polyclonal antibody to caspase-9 (p35/H170), and monoclonal antibody to β-actin (C-2) were from Santa Cruz (Santa Cruz, CA, USA). For immunohistochemistry studies, the monoclonal antibody to Ki67/MIB1 (catalog No. M7240) was purchased from Dako (Denmark). All antibodies were diluted in TBS-T (0.1% Tween-40 in 100 mM Tris-Cl, pH 7.4, 0.8% NaCl). Anti-BrdUdr (FITC; catalog No. 347583) was purchased from Becton Dickinson (NJ, USA); propidium iodide (PI) was from Roche (Basel, Switzerland), and all other chemicals were from Sigma-Aldrich. All cell culture reagents were purchased from Euroclone Life Sciences Division (Milano, Italy).
Cell cultures
The human colon cancer HCT116 cell line was obtained from the National Cancer Institute, NIH (Bethesda, MD, USA), and was adapted to propagate in RPMI 1640 medium supplemented with 5% heat-inactivated fetal calf serum, 2 mM L-glutamine and antibiotics. The cultures were grown in a humidified 37°C-incubator with 5% CO2 atmosphere.
In vitro cytotoxic activity of sclareol
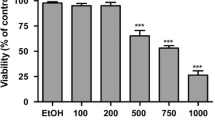
Cell viability was assessed at the beginning of each experiment by the trypan blue dye exclusion method, and was always greater than 95%. Cells were seeded into 96-well microtiter plates in 100 μL of medium at a density of 5000 cells per well, and subsequently, the plates were incubated at standard conditions for 24 h to allow the cells to resume exponential growth prior to addition of sclareol. Then, in order to measure the cell population, cells in one plate were fixed in situ with TCA followed by SRB staining, as described elsewhere [12]. To determine the sclareol activity, the compound dissolved in DMSO was added at 10-fold dilutions (from 100 to 0.01 μM) and incubation continued for an additional period of 48 h. The assay was terminated by addition of cold TCA followed by SRB staining and absorbance measurement at 530 nm, in an EL-311 BIOTEK microelisa reader (BioTek, Winooski, VT, USA), to determine the GI50, that is, the sclareol concentration required in the cell culture to inhibit cell growth by 50% as described [25].
Flow cytometry analysis of cell cycle and assessment of apoptosis
Untreated and sclareol-treated cells were collected at the desired periods of time, and then prepared and subjected to flow cytometry analysis to monitor perturbations in the cell cycle and apoptosis as described [18, 19, 26, 27]. In addition, apoptosis was assessed with the aid of the ssDNA Apoptosis, and the Cellular DNA Fragmentation ELISA kits. The ssDNA Apoptosis kit (Chemicon International, CA, USA) is used to detect formamide-denatured DNA in apoptotic cells with a monoclonal antibody that recognizes single-stranded DNA (ssDNA). In our studies, 5000 cells were seeded onto microplates and then treated with various sclareol concentrations for the desired periods of time, fixed into the wells, treated with formamide and stained with the antibody mixture as described in the assay protocol provided by the manufacturer. Further, the Cellular DNA Fragmentation ELISA kit (Roche, Switzerland) can quantify DNA fragments, after DNA damage, released in the cytoplasm upon induction of apoptosis. The experiments were carried out, as described by the manufacturer, in microtiter plates (containing 1 × 106 cells/ml) using various sclareol concentrations after BrdU-incorporation. At selected periods of scareol treatment, the supernatant was removed and kept at −20°C until use, and cell lysates and supernatants were assayed for presence of DNA fragments associated with apoptosis.
Protein extraction and Western blotting
Cells were washed twice in ice-cold PBS, detached with a latex cell-remover, collected by gentle centrifugation, and whole cell protein extracts were prepared as previously described [26–28]. Protein concentrations were determined with the Bradford assay, and subsequently, aliquots containing 30 μg of protein were subjected to gel electrophoresis on 8% or 10% polyacylamide SDS-gels, and then transferred to PVDF membranes (Millipore Immobilon). The membranes were stained with Ponseau S solution (0.1% Ponseau in 5% acetic acid; AlterChem, Athens, Greece), and proteins were visualized using an enhanced chemoluminescence detection system (ECL or ECL Plus, Amersham, Arlington Heights, IL) according to the manufacture's instructions.
Preparation of liposomes and liposomal sclareol
Liposomes and liposome-encapsulated sclareol (termed liposomal sclareol, hereafter) were prepared as described [13]. The encapsulated sclareol to lipid molar ratio was 0.43 suggesting an incorporation efficiency of nearly 80% [13]. Both, empty liposome (control) and liposomal sclareol suspensions were freeze-dried and stored at 4°C until used.
In vivo antitumor activity
Male SCID (NOD.CB17 Prkdcscid) mice were purchased from Jackson Laboratories/Charles River Laboratories (L’Arbresle, France). The mouse colony was maintained under restricted flora conditions in a pathogen-free environment in type IIL-cages. Male mice, 7–9 weeks old, were injected subcutaneously according to the British practice of bilateral trocar implants at the axillary region. Each inoculum contained 106 cells exponentially growing at the time of harvesting. The two tumor cell inoculums were added together for each mouse, thus producing a tumor burden per mouse value for data analysis. The advantage of the British system is reduced mouse-to-mouse variability and, thus, the ability to reduce the number of animals per group, with five being sufficient for accurate data analysis of tumor response and evaluation of toxicities. The virtues of the British system of mouse xenografting over the “traditional” single implantation are described in ref. [29]. The mice were randomly divided into groups of 6 to 8 animals per group. Treatments started when the average tumor volume had reached about 100 mm3. Tumor volume was calculated as described in ref. [29]. All administrations were intraperitoneal. Treated animals received a single injection daily for 5 days per week throughout the experiment. Tumor volume was measured with a calliper twice per week. In addition to tumor volume, we calculated the parameter, %ΔT/ΔC, where ΔT = T − D o and ΔC = C − D o (D o is the average tumor volume at the beginning of the treatment; T and C are the volumes of treated and untreated tumors, respectively, at a specified day). Concurrently, we scored the number of tumor-free animals, number of drug-related deaths, and average number of days required to reach a defined tumor volume. Optimal ΔT/ΔC value was used as a measure of drug activity. Losses of weight, neurological disorders and behavioral and dietary changes were also recorded as indicators of toxicity (side effects). The experiment was terminated when tumor size in untreated animals reached a volume of about 1000 mm3. All animals were treated according to Greek laws (2015/92), guidelines of the European Union and the European council (86/609 and ETS123, respectively), and Compliance with Standards for Human Care and Use of Laboratory Animals, NIH, USA (Assurance No. A5736-01).
Morphology and immunohistochemistry of tumors
Tumors were excised, fixed in buffered formalin (4%), embedded in paraffin, and 4-μm sections were prepared with the aid of a Leica, model RM2125, microtome. Subsequently, sections were stained with hematoxylin-eosin and examined under a microscope to assess the histological phenotype of the tumor, the type and degree of differentiation, and the presence of regressive changes. Other sections were de-paraffinized and subjected to standard two-stage indirect immunohistochemical technique (ABC method) using the polyclonal Ki67-antibody at 1:150 dilution and incubated overnight at room temperature. For antigen retrieval, the sections were pretreated (boiled in microwave oven) for 20 min in citrate buffer, pH 6 [30], and stained with the chromogen, DAB. Unstained and positively-stained nuclei were scored in 10 fields, from two independently-removed mouse biopsies (that is four tumors per group), at high power, and the percentage of the positively-stained cells was estimated. Sections stained with hematoxylin-eosin or examined for presence of Ki67 antigen were photographed under a Nikon Eclipse 80i equipped with Digital Sight DS-2Mv.
Statistical analysis
Significant difference in tumor volume was determined by the Student's t-test using the SPSS for Windows (release 11.0.0, SPSS Inc., USA) software package. A difference was considered significant if p < 0.05.

Sclareol-induced cell cycle perturbations. Identical asynchronous HCT116 cell cultures were treated with 100 μM sclareol for 8 h (histogram B), 16 h (histogram C) and 24 h (histogram D), and attached and detached cells were harvested and subjected to flow cytometry analysis of the relative DNA content (i.e., cell cycle perturbations and apoptosis). Control culture received carrier alone (histogram A). G1, G0 + G1 cells; G2, G2 + M cells; and, Ap, apoptotic cells (sub-G1 cell population). The estimate of each fraction expressed as % of the total cell population treated and as % of the gated live cells is shown in Table 1. Ten thousand events were scored per sample (histogram)
Results
Sclareol induces cell cycle arrest at G1-phase
The ability of sclareol to induce perturbations in the cell cycle was studied after we determined the GI50 to be 34 μM for HCT116 cells treated with various sclareol concentrations for 48 h. Subsequently, identical cultures of exponentially grown cells were treated with 100 μM sclareol for various periods of time for the flow cytometry studies. The results (histograms) of this study and the cell fractions (%) in each cell cycle phase and apoptosis are shown in Fig. 1 and Table 1. There were no significant changes in the cell cycle of the cells treated with sclareol for 8 h (see histograms A and B), but an increase in the G1- and Ap-fractions, at expense of the S-fraction, was observed at 16 h of treatment (histogram C) with a further increase in the Ap fraction after 24 h of treatment (histogram D). These results showed an increase in apoptosis but did not address whether the cells were at a specific cell cycle phase when entering apoptosis. Therefore to identify the cell cycle phase targeted by sclareol we studied the effect of this compound on cells already arrested at G1. This was accomplished by incubating several identical cell cultures for 72 h in media without serum (i.e., serumless media). At the end of the 72 h-incubation period the attached cells were gently washed with medium and afterwards received fresh medium containing 5% serum without or with 100 μΜ of sclareol. Exponentially growing HCT116 cells exhibited a typical distribution in various fractions (i.e., phases) of the cell cycle as shown in Fig. 2 (histogram A). This histogram indicates that approximately 34%, 39%, and 25% of the cells were in the G1-, S-, and G2-phase, repectively. However, after exposure of the cells for 72 h in serumless media, more than 80% of the cells were arrested in G1, while a small number of cells were still traversing the S- and G2-phases (Fig. 2, histogram B and Table 2). Subsequently, after the G1-arrested cells were transferred to media containing 5% serum for 12 h, about 50% of these cells had re-entered cycling as indicated by the decrease in the number of cells in G1, and an increase in the number of cells in the S-phase (see histogram C and Table 2). In contrast, the cells remained arrested in the G1-phase in the presence of sclareol in the serum supplemented media (histogram D). Further, at 24 h, cells in media containing serum without sclareol had progressed from the S- to the G2-phase (histogram E), whereas the presence of sclareol in the serum supplemented media continued to block progression of the cells from the G1- to S-phase (histogram F). Taken together, these results unequivocally demonstrated that the presence of sclareol in cultures of HCT116 cells led to arrest of these cells in the G1-phase of the cell cycle (Fig. 2, compare C to D and E to F).

Flow cytometry analysis of the cell cycle of serum-starved cells subsequently treated with sclareol. The histograms were derived from untreated exponentiall grown cells (A); cells grown in serumless medium for 72 h (B); cells grown in serumless media for 72 h, then receiving 5% serum for 12 h without (C) or with (D) 100 μM of sclareol; and cells grown in serumless media for 72 h, then receiving 5% serum for 24 h without (E) or with (F) 100 μM of sclareol. Ten thousand events were scored per sample (histogram) and estimate of each cell cycle phase is presented as percentage of the total cells treated in Table 2
Sclareol induces DNA damage in a time-dependent manner
The results of the flow cytometry studies demonstrated that in the presence of sclareol the HCT116 cells were arrested at the G1-phase of the cell cycle, whereas cells in other phases of the cell cycle died by apoptosis (Figs. 1 and 2). These results prompted us to further elucidate how early sclareol alters the conformation and integrity of the chromatin DNA. First, we monitored sclareol-induced conformational changes in the DNA using the ssDNA Apoptosis kit as described in Materials and Methods. The results of this study demonstrated that addition of 100 μM sclareol to HCT116 cell cultures resulted in presence of DNA breaks as early as 4 h after sclareol addition (Fig. 3, graph A). Further, chromatin DNA breaks continued to accumulate as the cells continued to be exposed to sclareol.

Sclareol-induced DNA damage is time-dependent. HCT116 cells were treated with 100 μM sclareol for various periods of time, and then, cells (cell lysates) were assayed for DNA unwinding (A) and presence of DNA fragments (B). Also, cell-conditioned media (supernatant) was assayed for presence of cell released DNA fragments (A). DIC microscopy (magnification 200×) of HCT116 cells treated for 24 h with carrier alone (C) and carrier with sclareol (D). Pc, positive control provided by the kit manufacturer; p ≤ 0.05
We also assessed the timing of appearance of DNA fragments in the cytoplasm and cell-conditioned media, after addition of sclareol to the cell culture, events that have been associated with late and irreversible stages of apoptosis. For this study, we utilized the Cellular DNA Fragmentation ELISA kit as described in Materials and Methods. The results indicated the absence of DNA fragments in cells treated for 4 h with 100 μM sclareol, but the presence of DNA fragments was readily detected at 8 h of sclareol treatment (Fig. 3, graph B). Further, the relative amount of DNA fragments continued to increase within the treated cells, whereas no DNA fragments were detected in the cell-conditioned media of the treated cells even after 24 h of treatment.
Microscopy observations of untreated and sclareol-treated HCT116 cells assessed that the treated cells had ceased to proliferate, were shrung and round, but retained their integrity (Fig. 3, panels C and D), that is, chromatin DNA changes and fragmentation preceded or concomitantly occurred with arrest of the cells at G1.
Activation of caspases in sclareol-treated HCT116 cells
Having established that sclareol induces DNA damage in a time-dependent manner, we sought to investigate whether sclareol treatment of HCT116 cells induces activation of caspase-8 and caspase-9 (Fig. 4). The results indicated that procaspase-8 was extensively processed at 8 h of sclareol treatment and almost completely processed at 16 h of treatment. On the other hand, procaspase-9 was also processed, although not as extensively as procaspase-8, at 8 h of treatment, but a nearly complete process had occurred at 16 h of treatment (Fig. 4).

Sclareol induces caspase activation and PARP cleavage. HCT116 cells were untreated (ut) or treated with 100 μM of sclareol for 4 h, 8 h, and 16 h and then, whole cell protein extracts were prepared and subjected to Western blot analysis for presence of procaspase-8, procaspase-9, procaspase-3, PARP and the control (housekeeping) protein, β-actin using specific antibodies
Consistent with the timing of activation of procaspases-8 and -9, processing of procaspase-3 was detectable at 8 h of sclareol treatment, whereas no procaspase-3 was detected at 16 h of sclareol treatment, apparently because it was fully converted, to active caspase-3. These observations were further confirmed upon monitoring the degradation of PARP, which is targeted by active caspase-3 [15–17]. The results again showed that the kinetics of PARP degradation was concurrent or closely followed the kinetics of procaspase-3 activation (Fig. 4). No degradation has been observed with a b-actin control.
Sclareol suppresses growth of HCT116 tumors
The ability of sclareol to inhibit HCT116 cell proliferarion followed by apoptosis prompted us to study the efficacy of this compound to inhibit growth of HCT116 tumors established as xenografts in immunodeficient SCID mice. For this study, we initially determined the MTD of sclareol administered intraperitoneally (ip) to the animals. In our hands, sclareol was found to be insoluble in water-containing solutions, and thus we chose to use a fine suspension of sclareol for ip administration. We tested several carriers (vehicles) in order to obtain a suspension meeting the following criteria: (i) the carrier alone is compatible with the host; (ii) the suspended sclareol does not precipitate after storage for an extensive period of time (nearly 24 h) at room temperature; and, (iii) the suspension can be well-tolerated after ip administration. These criteria were met with the use of a carrier consisting of normal saline (80%, v/v)—absolute ethanol (10%, v/v)—PEG 400 (5%, v/v)—Tween-40 (5%, v/v). We subsequently used this cocktail to determine that the MTD for sclareol alone (free sclareol) administered in SCID mice daily for 5 consecutive days was 50 mg/kg. Alternatively, sclareol was encapsulated into the lipid bilayer of liposomes as described elsewhere [13], and the MTD of the liposomal sclareol administered daily for 5 days was determined to be 275 mg/kg (data not shown).
To study the efficacy of free and liposomal sclareol in suppressing tumor growth, 30 SCID mice were xenografted (2 xenografts per mouse) with HCT116 cells, and when the tumors reached a size of 100 mm3, animals were divided to 5 groups (6 mice per group). One group of mice received no further treatment (control #1), whereas the other groups received vehicle+free sclareol (50 mg/kg), vehicle alone (control #2), vehicle + empty liposomes (control #3), and vehicle+liposomal sclareol (275 mg/kg). Each treatment consisted of 3 weekly cycles, each cycle consisting of 5 consecutive days of compound administration per week. The study was terminated, when the average tumor size of the untreated animals reached approximately 1000 mm3 (end-point). Following termination of tumor measurements in untreated and treated mice, we utilized the average tumor measurements to calculate %ΔT/ΔC as a function of time (Fig. 5). The first observation was that free sclareol (at 50 mg/kg) had virtually no effect on the tumor size (Fig. 5(A)). Similarly, administration of vehicle + empty liposomes had no effect on tumor size. However, administration of vehicle + liposomal sclareol (at 275 mg/kg) resulted in a dramatic suppression of tumor growth (Fig. 5(A)). In fact, during the initial period of the liposomal sclareol treatment the tumor size suppression was about 50% followed by 40% suppression at later periods of treatment (Fig. 5(A)). No side effects (drug related deaths, loss of weight, neurological disorders etc) have been recorded during the treatment with the MTDs of free and liposomal sclareol. Therefore, taken together the results demonstrate that (a) the MTD of liposomal sclareol is more than 5-fold higher than the MTD of free sclareol; and (b) the antitumor activity of sclareol is exhibited at doses that can be administered only after this compound is encapsulated in liposomes.

Growth of HCT116 tumors in mice treated with sclareol and liposomal sclareol. (A), average tumor size (in mm3) of each mouse group (6 mice per group that is 12 tumors per group) as a function of time (in Days); and, (B), %ΔT/ΔC as a function of time (in Days) calculated from the measurements in A for untreated mice (•) and mice receiving free sclareol (50 mg/kg, ▪), empty liposomes (○), and liposomal sclareol (275 mg/kg, ▴). Treatment was for three cycles, each cycle consisting of five consecutive days of compound administration followed by two consecutive days with no treatment. Statistical evaluation was assessed with the two-tailed t-test. Points with p ≤ 0.05 are indicated by a star (*)
Characteristics of sclareol-treated tumors
To address the ability of sclareol to induce tumor growth suppression in vivo we first examined by direct observations the growth of tumors in the mice 30 days post-inoculation of the tumor cells. Advanced and similar tumor growth was readily observed in mice that received no treatment or treatment with sclareol or empty liposomes alone, but tumor growth was substantially slower in mice treated with liposomal sclareol (mice with tumors not shown). We then observed the microscopic morphology of the sclareol-treated tumor in hematoxylin-eosin-stained sections (Fig. 6). A photograph of a representative section shows a highly aggressive undifferentiated carcinoma with extensive foci of necrosis (Fig. 6(A)). Further, we investigated whether sclareol treatment affects proliferation of cancer cells in the section by detecting the presence of the nuclear Ki67/MIB1 protein, a marker indicative of active cell proliferation (reviewed in refs. [30–33]). More than 80% of the cells stained positive for Ki67 in the untreated tumor (Fig. 6(B)) and tumors treated with liposomes (Fig. 6(C)) alone or free-sclareol (not shown) indicating abundant presence of the Ki67 antigen, that is, the cancer cells were actively proliferating. In contrast, liposomal sclareol treatment resulted in a 50% reduction of Ki67 staining in the tumor. These observations taken together demonstrated that treatment with liposomal sclareol had impaired the ability of tumor cells to proliferate.

Detection of expression of the cell proliferation-associated protein, Ki67, in tumor sections. At the end of each treatment, the animals were euthanized, the tumors were surgically removed from the animals and embedded in paraffin and 4-μm sections were observed under a microscope. (A), highly aggressive undifferentiated carcinoma with extensive foci of necrosis (hematoxylin-eosin stain, 400× magnification); (B), intensive nuclear expression of Ki67/MIB1 in untreated tumor [85±11% positive cells; ABC-stain; 400 × magnification), (C), expression of Ki67/MIB1 in tumors treated with empty liposomes (percentage 80±8% positive cells) and (D), loss of Ki67/MIB1 expression after treatment with liposomal sclareol [(50 ± 10% positive cells, p ≤ 0.05)]
Discussion
There is increasing experimental and epidemiological evidence that plant-derived chemicals (phytochemicals) possess the potential to serve as chemopreventive agents that arrest or reverse carcinogenesis (for recent reviews see refs. [34–38]). In this regard, we have previously provided experimental findings to demonstrate that the phytochemical diterpene, sclareol, exhibits anticancer activities in vitro [11, 12] and in vivo [13]. In the initial reports, we demonstrated that sclareol induced apoptosis in human leukemia cell lines associated with down-regulation of the anti-apoptotic gene, c-myc, in absence of quantitative changes in the pro-apoptotic protein, Bcl-2 [9], and that apoptosis was independent on p53 expression in human breast cancer cell lines [12].
In this report, we have extended our previous studies to show that sclareol can induce arrest at the G1-phase of the cell cycle in human colon cancer HCT116 cells. Interestingly, G1-arrested cells, following serum starvation, were not killed by sclareol, whereas, the same compound killed cells not arrested at G1. This finding suggests that sclareol can distinguish between non-proliferating and proliferating cells, that is, sclareol possesses a property highly desirable for an anti-cancer drug. Further, our studies have shown that sclareol-induced apoptosis is associated with activation of caspases-8 and -9, implying that, sclareol-induced cell death is associated with activation of both mitochondrion and death receptor apoptosis pathways, followed by activation of caspase-3 and subsequent degradation of the protein PARP. Activation of caspase-8 or -9 is an early event required to trigger the death receptor-dependent and mitochondrion-dependent apoptosis pathways, respectively, whereas, activation of caspase-3 and subsequent PARP degradation are late events in the apoptosis pathways [15–17]. These results are consistent with a report that both apoptotic pathways are triggered in human leukemia cell lines treated with other labdane diterpenes [10]. However, activation of caspases-8 and -9 does not exclude the possibility that other caspase-independent mechanisms of cell death are executed by proteases other than caspases [39, 40]. In fact, we have recently shown that such a variant form of apoptosis display only some characteristics typical of the “classical” apoptosis pathways in HCT116 cells treated with the plant-derived product, mastic gum [18, 19]. At any rate, we have concluded that sclareol-induced apoptosis in HCT116 cells is associated with biochemical processing of cellular macromolecules, but we have not investigated the form of apoptosis induced. It should also be noted that treatment of HCT116 cells with sclareol for 8 h irreversibly induced apoptosis even after transferring the cells to media without sclareol (results not shown) indicating that once sclareol initiated the activation of the apoptotic mechanism, continuous exposure of the cells to sclareol was not required for apoptosis to be completed.
Subsequently, we evaluated the antitumor efficacy of sclareol in vivo using the human HCT116 tumor xenograft/SCID mouse model. In preliminary experiments, we determined that the high insolubility of the lipophilic sclareol in aqueous solutions (carriers) was limiting us to administer into mice doses lower than 50 mg/kg, applying a schedule of a daily administration for five consecutive days. Even a single administration of a dose higher that 50 mg sclareol per kg was adequate to generate extensive toxicity and eventually death to the animals [13]. However, a dose of 50 mg/kg had no effect on the progression (growth) of the HCT116 tumors grown on the mice. Therefore, we alternatively administered sclareol encapsulated into specifically prepared liposomes [13]. Liposomes can be used as carriers for delivery of a variety of compounds at higher doses, to elicit improved pharmacokinetic properties and pharmacological responses and reduce toxicity observed in systemically administered drugs alone [20–24, 41, 42]. Specifically for anticancer drugs, liposomes tend towards a preferential accumulation in tumor sites and an increased uptake by cells via the enhanced permeability retention (EPR) effect [43].
Our liposomal sclareol preparations allowed us to administer a daily dose of 275 mg sclareol/kg, that is, a dose much higher than 50 mg/kg of the sclareol in aqueous carrier. Administration of a dose of empty (control) liposomes equal to the quantity of the sclareol-containing liposomes indicated absence of apparent toxicity in the mice assuring that the experimental results observed or measured were due to the encapsulated sclareol rather than to the carrier (liposomes). In fact, at the endpoint of the experiment (i.e., when the average tumor size was approximately 1000 mm3 in the control animals), the average size of tumors in the treated animals was about 50% lower than in control animals.
The dramatic suppression of HCT116-tumor growth after sclareol treatment could be due to sclareol-inhibited proliferation or apoptotic death of the tumor cells as observed in the in vitro studies. To investigate this possibility, we stained tumor sections form treated or untreated animals for expression of the nuclear antigen, Ki67/MIB1, a marker selectively expressed in actively proliferating cells [30–33]. Our results clearly show that liposomal sclareol inhibits expression of Ki67 indicating a direct effect on cell proliferation.
In general, other members of the labdane diterpene class have exhibited significant apoptotic activity against human cancer cells in vitro [10, 11, 44, 45], but still very little is known about the anticancer properties of these compounds. Moreover, the results described in this report have demonstrated for the first time the ability of a labdane diterpene, sclareol, to suppress growth of human colon cancer in vivo. It is our assumption that sclareol, and perhaps other labdane diterpenes, can also suppress or regress the growth of other human tumors, in addition to HCT116 colon tumor, in the human xenograft/mouse model. This assumption is currently tested by our group. In conclusion, the results presented in this report warrant further investigations on the anticancer activity of sclareol and the possibility that this compound can be developed to a novel anticancer drug.
References
Connolly JD, Hill RA (1991) Dictionary of terpenoids. Chapman & Hall, London
Demetzos K, Harvala C, Philianos SM, Skaltsounis AL (1990) A new labdane type diterpene and other compounds from the leaves of Cistus creticus (L) Heywood. J Nat Prod 53:1315–1368
Demetzos C, Stahl B, Anastassaki T, Gazouli M, Tzouvelekis, L, Rallis M (1999) Chemical analysis and antimicrobial activity of the resin ladano, of its essential oil and of the isolated compounds. Planta Med 65:76–78
Demetzos C, Dimas K (2001) Labdane-type diterpenes: Chemistry and biological activity. In: Atta-ur-Rahman (ed) Studies in natural products chemistry: Bioactive natural products (Part F). Oxford, Elsevier Science, pp 235–292
Seamon KB, Daly JW (1981) Activation of adenylate cyclase by the diterpene forskolin does not require the guanine nucleotide regulatory protein. J Biol Chem 256:9799–9802
Dimas K, Demetzos C, Marsellos M, Sotiriadou R, Malamas M, Kokkinopoulos D (1998) Cytotoxic activity of labdane type diterpenes against human leukemic cell lines in vitro. Planta Med 64:208–211
Dimas K, Demetzos C, Mitakou S, Vaos B, Marselos M, Tzavaras T, Kokkinopoulos D (1999) Cytotoxic activity and antiproliferative effects of a new semi-synthetic derivative ent-3β-OH-manoyl oxide, on human leukemic cell lines. Anticancer Res 19:4065–4072
Dimas K, Kokkinopoulos D, Demetzos C, Vaos B, Marselos M, Malamas M, Tzavaras T (1999) The effect of sclareol on growth and cell cycle progression of human leukemic cell lines. Leuk Res 23:217–234
Dimas K, Demetzos C, Ioannidis P, Trangas T (2001) Labdane type diterpenes down-regulate the expression of c-Myc protein but not of Bcl-2, in human leukemia T-cell lines undergoing apoptosis. Leuk Res 25:449–454
Souza-Fagundes EM, Brumatti G, Martins-Filho OA, Correa-Oliveira R, Zani CL, Amarante-Mendesr GP (2003) Myriadenolide, a labdane diterpene isolated from Alomia myriadenia (asteraceae) induces depolarization of mitochondrial membranes and apoptosis associated with activation of caspases-8, -9, and -3 in Jurkat and THP-1 cells. Exp Cell Res 290:420–426
Li WX, Cui CB, Cai B, Yaor XS (2005) Labdane-type diterpenes as new cell cycle inhibitors and apoptosis inducers from Vitex trifolia L. J Asian Nat Prod Re 7:95–105
Dimas K, Papadaki M, Tsimplouli C, Hatziantoniou S, Alevizopoulos K, Pantazis P, Demetzos C (2006). Labd-14-ene-8, 13-diol (sclareol) induces cell cycle arrest and apoptosis in human breast cancer cells and enhances the activity of anticancer drugs. Biomed Pharmacother 60:127–133
Hatziantoniou S, Dimas K, Georgopoulos A, Sotiriadou N, Demetzos C (2006) Cytotoxic and antitumor activity of liposome-incorporated sclareol against cancer cell lines and human colon cancer xenografts. Pharmacol Res 53:80–87
Ulubelen A, Topcu G, Eris C, Sonmez U, Kartal M, Kurucu S, Bozok-Johansson C (1994) Terpenoids from Salvia sclarea. Phytochemistry 36:971–974
Strasser A, O’Connor L, Dixit VM (2001) Apoptosis signaling. Ann Rev Biochem 69:217–245
Salvase GS, Abrams JM (2004) Caspase activation—stepping on the gas or releasing the brakes? Lessons from humans and flies. Oncogene 23:2774–2784
Krammer PH, Debatin KM (2004) Death receptors in chemotherapy and cancer. Oncogene 23:2950–2966
Balan KV, Demetzos C, Prince J, Dimas K, Cladaras M, Han Z, Wyche JH, Pantazis P (2005) Induction of apoptosis in human colon cancer HCT116 cells treated with an extract of the plant product, Chios mastic gum. In Vivo 19:93–102
Balan KV, Prince J, Han Z, Dimas K, Cladaras M, Wyche JH, Sitaras NM, Pantazis P (2006) Antiproliferative activity and induction of apoptosis in human colon cancer cells treated in vitro with constituents of a product derived from Pistacia lentiscus L. var. chia. Phytomedicine, in press
Kshirsagar NA, Pandya SK, Kirodian BG, Sanath S (2005) Liposomal drug delivery system from laboratory to clinic. J Postgrad Med 51:S5–S15
Zamboni WC (2005) Liposomal, nanoparticle, and conjugated formulations of anticancer agents. Clin Cancer Res 11:8230–8234
Hart SL (2005) Lipid carriers for gene therapy. Curr Drug Deliv 2:423–428
Chen Y, Dalwadi G, Benson HA (2004) Drug delivery across the blood-brain barrier. Curr Drug Deliv 1:361–376
Bartsch M, Weeke-Klimp AH, Meijer DK, Scherphof GL, Kamps JA (2005) Cell-specific targeting of lipid-based carriers for ODN and DNA. J Liposome Res 15:59–92
Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren J, Bokesch H, Kenney S, Boyd M (1990) New colorimetric cytotoxicity assay for anticancer drug screening. J Natl Cancer Inst 82:1107–1114
Pantazis P, Chatterjee D, Han Z, Wyche JH (2000) Differentiation of human malignant melanoma cells that escape apoptosis following treatment with 9-nitrocamptothecin in vivo. Neoplasia 1:231–240
Hu X, Balan KV, Ramos-DeSimone N, Wyche JH, Han Z, Pantazis P (2003) Differential susceptibility to 9-nitrocamptothecin (9-NC)-induced apoptosis in clones derived from a human ovarian cancer cell line, possible implications in the treatment of ovarian cancer patients with 9-NC. Anticancer Drugs 14:427–436
Corbett T, Valeriote F, LoRusso P, Polin L, Pancharov C, Pugh S, Whyte K, Knight J, Demchick L, Jones J, Jones L, Lissow L (1997) In vivo methods for screening and preclinical testing. In vivo methods for screening and preclinical testing. In: Teicher B (ed) Anticancer drug development guide: Preclinical screening, clinical trials, and approval. Humana Press, Tobwa, NJ, pp 75–99
Pluquet O, North S, Bhoumik A, Dimas K, Ronai Z, Hainaut P (2003) The cytoprotective aminothiol WR1065 activates p53 through a non genotoxic signaling pathway involving c-Jun N-terminal kinase. J Biol Chem 278:11879–11887
Cattoretti G, Becker MH, Key G, Duchrow M, Schluter C, Galle J, Gerdes J (1992) Monoclonal antibodies against recombinant parts of the Ki-67 antigen (MIB 1 and MIB 3) detect proliferating cells in microwave-processed formalin-fixed paraffin sections. J Pathol 168:57–363
Yu CC, Woods AL, Levison DA (1992) The assessment of cellular proliferation by immunohistochemistry, a review of currently available methods and their applications. Histochemistry J 24:121–131
Magdelenat H (1992) Tumour markers in oncology: past, present and future. J Immunol Methods 150:133–143
van Diese PJ, Van Der Wall E, Baak JP (2004) Prognostic value of proliferation in invasive breast cancer: a review. J Clin Pathol 57:675–681
Singh RP, Agarwal R (2006) Natural flavonoids targeting deregulated cell cycle progression in cancer cells. Curr Drug Targets 7:345–354
D’Incalci M, Steward WP, Gescher AJ (2005) Use of cancer chemopreventive phytochemicals as antineoplastic agents. Lancet Oncol 6:899–904
Nishino H, Murakoshi M, Mou XY, Wada S, Masuda M, Ohsaka Y, Satomi Y, Jinno K (2005) Cancer prevention by phytochemicals. Oncology 69(suppl 1):38–40
Collins AR (2005) Antioxidant intervention as a route to cancer prevention. Eur J Cancer 41:1923–1930
Gescher A (2004) Polyphenolic phytochemicals versus non-steroidal anti-inflammatory drugs: which are better cancer chemopreventive agents? J Chemother 16(suppl 4):3–6
Jäättelä M (2004) Multiple cell death pathways as regulators of tumor initiation and progression. Oncogene 23:2746–2756
Lockshin RA, Zakeri Z (2004) Caspase-independent cell death? Oncogene 23:2766–2773
Allen TM, Cullis RP (2004) Drug delivery systems: entering the mainstream. Science 19:1818–1822
Batist G, Ramakrishnan G, Rao CS, Chandrasekharan A, Gutheil J, Guthrie T, Shah P, Khojasteh A, Nair MK, Hoelzer K, Tkaczuk K, Park YC, Lee LW (2001) Reduced cardiotoxicity and preserved antitumor efficacy of liposome-encapsulated doxorubicin and cyclophosphamide compared with conventional doxorubicin and cyclophosphamide in a randomized, multicenter trial of metastatic breast cancer. J Clin Oncol 19:1444–1454
Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK (1995) Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res 55:3752–3756
Jung M, Ko I, Lee S, Choi SJ, Youn BH, Kim SK (1998) A concise synthesis and in vitro cytotoxicity of new labdane diterpenes. Bioorg Med Chem Lett 8:3295–3298
Lee SO, Choi SZ, Choi SU, Lee KC, Chin YW, Kim J, Kim YC, Lee KR (2005) Labdane diterpenes from Aster spathulifolius and their cytotoxic effects on human cancer cell lines. J Nat Prod 68:1471–1474
Acknowledgments
Authors would like to thank Dr. Stamatis Pagakis and Dr. Eleni Rigana of the Biological Imaging Unit of the Foundation for Biomedical Research of Academy of Athens, for their contribution and help in microsopy studies.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dimas, K., Hatziantoniou, S., Tseleni, S. et al. Sclareol induces apoptosis in human HCT116 colon cancer cells in vitro and suppression of HCT116 tumor growth in immunodeficient mice. Apoptosis 12, 685–694 (2007). https://doi.org/10.1007/s10495-006-0026-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-006-0026-8