
Abstract
Alpha-lipoic acid (LA), which plays a pivotal role in mitochondrial energy metabolism, is an endogenous dithiol compound with an array of antioxidative functions. It has been shown that LA triggers cell death in tumor cell lines, whereas non-transformed cells are hardly affected. In the present study, we analyzed the cytotoxicity of LA on colorectal cancer (CRC) cells differing in their p53 status and investigated a putative synergistic effect with the anticancer drug 5-fluorouracil (5-FU). We show that LA induces a dose-dependent decrease in cell viability, which was independent of the p53 status as attested in isogenic p53-proficient and p53-deficient cell lines. This effect was largely attributable to cell death induction as revealed by Annexin-V/PI staining. LA-treated HCT116 cells underwent caspase-dependent and caspase-independent cell death, which was blocked by the pan-caspase inhibitor zVAD and the RIP-kinase inhibitor Necrostatin-1, respectively. In CaCO-2 and HT29 cells, LA induced caspase-dependent cell demise via activation of caspase-9, caspase-3 and caspase-7 with subsequent PARP-1 cleavage as demonstrated by immunoblot analysis, activity assays and pan-caspase inhibition. Interestingly, LA treatment did neither activate p53 nor induced genotoxic effects as shown by lack of DNA strand breaks and phosphorylation of histone 2AX. Finally, we provide evidence that LA increases the cytotoxic effect induced by the anticancer drug 5-FU as revealed by significantly enhanced cell death rates in HCT116 and CaCO-2 cells. Collectively, these findings demonstrate that LA induces CRC cell death independent of their p53 status and potentiates the cytotoxicity of 5-FU without causing DNA damage on its own, which makes it a candidate for tumor therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alpha-lipoic acid (LA) is a dithiol compound synthesized de novo in mitochondria, where it is covalently bound to multienzyme complexes such as pyruvate dehydrogenase, representing a pivotal cofactor for decarboxylation of α-keto acids (Shay et al. 2009). Dietary supplementation with LA results in its gastrointestinal absorption (Carlson et al. 2007; Teichert et al. 1998). The uptake of LA into tissues and cells is mediated by diverse mechanisms, involving the monocarboxylate transporter (Takaishi et al. 2007) and Na+-dependent multivitamin transporter (Prasad et al. 1998). After internalization, LA is converted into its reduced form dihydrolipoic acid (DHLA) in a NADPH-dependent manner, most likely catalyzed by thioredoxin reductase (Handelman et al. 1994; Jones et al. 2002). Owing to the chemical reactivity of its dithiolane ring, LA and its reduced counterpart DHLA form a potent redox couple and display an impressive array of antioxidant functions (Rochette et al. 2013). Both compounds were reported to directly scavenge reactive oxygen species (ROS) and inhibit their formation by chelating transition metal ions including Fe2+. In addition, LA was shown to enhance GSH levels in various cellular systems (Rochette et al. 2013), which was mediated by the redox-sensitive transcription factor Nrf2 (Suh et al. 2004). Based on these ROS-defending properties, LA bears great potential for the treatment of chronic diseases associated with high levels of oxidative stress, such as diabetes mellitus (Smith et al. 2004). Noteworthy, LA was also shown to reduce inflammation and oxidative stress in a mouse model of chemically induced colitis (Trivedi and Jena 2013).
Beside this well-described function, a growing body of evidence shows that LA is capable of suppressing growth of various tumor cell lines, interfering with the cellular redox state. It has been reported that LA treatment of H460 human lung cancer cells resulted in increased ROS formation at higher doses, which triggered caspase-dependent cell death by down-regulation of the antiapoptotic factor Bcl-2 (Moungjaroen et al. 2006). In line with these findings, LA also increased ROS levels in non-small cell lung cancer cells associated with a pronounced inhibition of cell proliferation and suppressed the activation of Met tyrosine kinase (Michikoshi et al. 2013). The pro-apoptotic effects of LA were confirmed in MCF7 breast cancer cells, where it elicited p27Kip1-dependent cell cycle arrest and subsequent apoptosis (Dozio et al. 2010). Further studies demonstrated that LA also triggers cell death in HepG2 hepatoma and Jurkat T-lymphoma cells (Simbula et al. 2007; van de Mark et al. 2003). Cell death induction by LA was exclusively observed in tumor cell lines, whereas primary cells such as mitogen-activated human peripheral blood lymphocytes, non-transformed human epidermal fibroblasts and colonocytes were little affected by LA and displayed only moderate growth arrest (Pack et al. 2002; van de Mark et al. 2003; Wenzel et al. 2005).
Colorectal cancer (CRC) is one of the most frequent tumors worldwide and involves several dietary and non-dietary risk factors (Fahrer and Kaina 2013). Its therapy requires a multimodality approach, which comprises surgical resection of the tumor followed by chemotherapy and/or radiation therapy (Cunningham et al. 2010). The chemotherapy is based on the pyrimidine analogue 5-fluorouracil (5-FU) that, depending on the tumor stage, is combined with the platinum agent oxaliplatin or the topoisomerase inhibitor irinotecan (Cunningham et al. 2010). Despite substantial progress in the therapy of advanced CRC, 5-year survival rates account for only 10 % (Kopetz et al. 2009), highlighting the need for improved therapies and new concepts that include targeted disruption of cancer cell metabolism and interference with the redox state of cancer cells (Gogvadze et al. 2009; Hanahan and Weinberg 2011).
Given the impact of LA on cellular redox balance and its pivotal role in mitochondrial energy metabolism, we hypothesized that LA is effective in CRC cells and may synergize with the antimetabolite 5-FU, used as “gold standard” in CRC therapy. To this end, the growth inhibitory and cytotoxic effects of LA were assessed with respect to the cellular p53 status. Furthermore, LA-induced cell death pathways were analyzed, revealing caspase-dependent and caspase-independent cell demise. To rule out genotoxic effects, LA-mediated generation of DNA strand breaks was monitored by different methods, showing that LA acts in a DNA damage-independent manner. Finally, a combination of 5-FU and LA was tested in CRC cells and demonstrated a significant increase in 5-FU-mediated cytotoxicity.
Materials and methods
Material
Staurosporine and R (+) alpha-lipoic acid (LA) were purchased from Sigma (Deisenhofen, Germany). Doxorubicin and 5-fluorouracil (5-FU) were obtained from Medac (Wedel, Germany). Antibody against phosphorylated histone 2AX was from Abcam (Cambridge, UK), and antibodies against cleaved caspase-3, cleaved caspase-9 and caspase-8 were from Cell Signaling (Denver, USA). The antibodies against Hsp90, caspase-2, caspase-3, caspase-9, p21 and p53 were obtained from Santa Cruz Biotechnology (Heidelberg, Germany). PARP-1 antibody was a kind gift of Prof. Dr. Alexander Bürkle (University of Konstanz, Germany). Necrostatin-1 was received from Enzo Life Sciences (Lausanne, Switzerland), zVAD was from R&D Systems (Wiesbaden, Germany) and Olaparib was from Selleck (Houston, USA). Cell culture medium and supplements were obtained from Life Technologies (Darmstadt, Germany). Standard chemicals were purchased at Sigma (Deisenhofen, Germany) unless otherwise stated.
Cell culture and treatments
The isogenic colon carcinoma cell lines HCT116-p53+/+ and HCT116-p53−/− were obtained from Prof. Dr. Bert Vogelstein (Baltimore, USA). CaCO-2 cells were a kind gift of Dr. Cornelia Dietrich (Department of Toxicology, University Medical Center Mainz). HT29 cells were obtained from Prof. Dr. Gerhard Fritz (Institute of Toxicology, University Medical Center Düsseldorf). HCT116 cells were maintained in DMEM supplemented with 10 % FCS and antibiotics (100 U/mL penicillin and 100 µg/mL streptomycin). CaCO-2 cells were cultured in MEM with 10 % FCS, antibiotics and nonessential amino acids. HT29 cells were maintained in RPMI 1640 medium containing 10 % FCS and antibiotics. Cells were routinely trypsinized and reseeded three times per week.
LA was prepared as 200 mM stock solution in 100 % ethanol. LA was directly added to cells in fresh medium and incubated for the time points indicated. As solvent control, ethanol was used corresponding to the respective LA dose. The pan-caspase inhibitor zVAD was dissolved in DMSO as 20 mM stock solution. Cells were supplemented with 20 µM zVAD 24 and 48 h following LA treatment. The RIP1-kinase inhibitor Necrostatin-1 was prepared in DMSO (77 mM stock) and added to LA-treated cells after 24 and 48 h with a final concentration of 50 µM. Olaparib was also dissolved in DMSO (20 mM stock), and cells were treated as described above at a final dose of 5 µM. Staurosporine was dissolved in DMSO (1 mM stock) and used at a concentration of 1 µM. All inhibitors were stored as aliquots at −20 °C and used freshly.
Determination of cell viability by ATP assay
To analyze the effects of LA on the viability of different cell lines, cells were grown in white 96-well plates overnight (HCT116-p53+/+: 5 × 103 cells/well; HCT116-p53−/−: 6.5 × 103 cells/well; HT29: 7.5 × 103 cells/well; and CaCO-2: 7.5 × 103 cells/well) and incubated with increasing LA concentrations (100–4,000 µM, depending on cell line) for 72 h. In addition, 5-FU was used (5 µM or 10 µM as indicated). Cell viability was then determined with CellTiter-Glo®Luminescent Cell Viability Assay (Promega, Mannheim, Germany) according to the manufacturer’s instructions using MicroLumat Plus 96-well plate reader (Berthold, Bad Wildbad, Germany). IC50-values were determined by GraphPad Prism 5.0 software. To this end, LA concentrations were transformed into the log scale and plotted against the cell viability, and the curve was fitted by nonlinear regression with variable slope (see supporting information, Figure S1).
Assessment of cell morphology by light microscopy
Cells were grown overnight (HCT116-p53+/+: 1 × 105 cells/dish and CaCO-2: 1.5 × 105 cells/dish) on CELLview™ cell culture dishes (Greiner, Frickenhausen, Germany) and treated with LA or ethanol as solvent control for 72 h. Cell morphology was monitored with a Zeiss Axio Observer.Z1 microscope equipped with a AxioCam MRc (Zeiss, Oberkochen, Germany). Images were acquired with Zen software (Zeiss) and processed by Image J (NIH, USA).
Flow cytometry-based measurement of cell death frequency and subG1 induction
Cell death rate was determined by Annexin-V/PI staining as described previously (Fahrer et al. 2014). Briefly, cells seeded in 6-well plates (HCT116-p53+/+: 1 × 105 cells/well; HCT116-p53−/−: 1.3 × 105 cells/well; and CaCO-2: 1.5 × 105 cells/well) were challenged with LA (500 up to 2,000 µM) and incubated for 72 h. As negative control, cells were treated with 1 % ethanol (corresponding to 2,000 µM LA). Unattached and attached cells were harvested, washed in PBS and then resuspended in Annexin-V-binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2, 0.1 % BSA) supplemented with Annexin-V-FLUOS (Roche Diagnostics, Mannheim, Germany). After incubation on ice for 15 min, propidium iodide (PI) (Sigma-Aldrich, Munich, Germany) was added to a final concentration of 1 µg/mL, and cells were analyzed by flow cytometry using FACSCanto II (Becton–Dickinson, Heidelberg, Germany). Following gating of cells, early apoptotic (Annexin-V positive) and late apoptotic/necrotic cell populations (Annexin-V and PI-positive) were determined by FACSDiva software.
For cell cycle analysis, cells were harvested and washed twice in PBS. Following ethanol precipitation at −20 °C for 1 h, pellet was resuspended in PBS supplemented with RNase A (20 µg/mL) and incubated for 1 h at RT. Subsequently, PI was added to a final concentration of 10 µg/mL, and cells were analyzed for DNA content by flow cytometry using FACSCanto II. Cell cycle distribution and subG1 population were analyzed with Flowing Software 2.5.1 (Perttu Terho, Turku Centre for Biotechnology, Finland).
Measurement of mitochondrial membrane potential
Cells were grown in 6-cm dishes (HCT116-p53+/+: 1 × 105 cells/dish and HCT116-p53−/−: 1.3 × 105 cells/dish) and treated with 1 mM LA for 72 h. As negative controls, cells were left untreated of incubated with the solvent control ethanol. Valinomycin served as positive control. Mitochondrial membrane potential was assessed with the cationic, lipophilic dye JC-1 (5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzamidazolylcarbocyanine iodide) that enters mitochondria in an electrochemical potential-dependent manner. Due to its mitochondrial accumulation, JC-1 forms red fluorescent aggregates. Upon depolarization of mitochondria, JC-1 is distributed throughout the cytosol, resulting in JC-1 monomers that exhibit green fluorescence. To this end, cells were harvested and stained with JC-1 for 20 min according to the manufacturer’s protocol. Subsequently, red (585 nm) and green (530 nm) fluorescence emission was monitored by flow cytometry using a FACSCanto II cytometer and evaluated by FACSDiva software.
Analysis of DNA fragmentation by agarose gel electrophoresis
To assess DNA fragmentation as a hallmark of apoptosis, HCT116 cells seeded in 6-cm dishes (1 × 105 cells/dish) were treated with 1 mM LA for 72 h. STS served as positive control. Dislodged and attached cells were harvested by centrifugation, resuspended in lysis buffer (0.1 % Triton X-100, 5 mM Tris–HCl pH 8.0, 20 mM EDTA) and then supplemented with PEG-8000 solution (40 % PEG 800, 1 M NaCl). After lysis and centrifugation, the supernatant was mixed 1:1 with phenol:chloroform:isoamyl alcohol (Roth, Karlsruhe, Germany). Following centrifugation, DNA was precipitated by addition of 100 % ethanol at −80 °C for 1 h. DNA was washed with 70 % ethanol and dried at 55 °C. Pellets were resuspended in TE buffer (Tris 10 mM, EDTA 1 mM, pH 8.0) containing RNase A (200 µg/mL) and incubated for 1 h at 37 °C. DNA concentration and purity were determined using a NanoDrop 2000 (Thermo Scientific, Dreieich, Germany), and samples were loaded on a 1.5 % agarose gel containing ethidium bromide. DNA was separated at 20 V for 7 h and finally visualized by an InGenius (Syngene, Cambridge, UK) system equipped with a QICAM12-bit camera (QImaging, Surrey, Canada).
Caspase-3/7 and Caspase-9 activity assay
To further analyze the involvement of apoptosis signaling, the activation of the initiator caspase-9 and executioner caspase-3 and caspase-7 was assessed by means of the Caspase-Glo® 9 Assay and the Caspase-Glo® 3/7 Assay (Promega, Mannheim, Germany), respectively. To this end, cells seeded in white 96-well plates (HCT116-p53+/+: 5 × 103 cells/well; HT29: 7.5 × 103 cells/well; and CaCO-2: 7.5 × 103 cells/well) were treated with increasing LA concentrations (250–2,000 µM, depending on cell line). Staurosporine (STS), a strong inducer of apoptosis, was included as a positive control. After 72 h, cells were lysed and a luminogenic caspase-3/7 or caspase-9 substrate was added according to the manufacturer’s instructions. For detection of caspase-9 activity, the substrate was supplemented with the proteasome inhibitor MG-132 (Promega, Mannheim, Germany) to reduce background proteolysis. Following an incubation period of 30 min, luminescence was determined in a MicroLumat Plus 96-well plate reader (Berthold, Bad Wildbad, Germany).
Determination of ROS formation
Cells were grown in 6-well plates overnight (HCT116-p53+/+: 3 × 105 cells/well and CaCO-2: 1.5 × 105 cells/well) and then incubated with two different LA concentrations (200 and 2,000 µM) for 24 h. As positive control, cells were challenged with hydrogen peroxide (100 and 400 µM, respectively) for 20 min. Subsequently, cells were washed twice with PBS and incubated with the ROS sensor CM-H2DCFDA (Life Technologies, Darmstadt, Germany) for 30 min. Cells were washed, trypsinized and pelleted by centrifugation. Cells were then resuspended in PBS and analyzed by flow cytometry using FACSCanto II (Becton–Dickinson, Heidelberg, Germany). Mean fluorescence intensity was assessed by FACSDiva software (Becton–Dickinson, Heidelberg, Germany).
Alkaline Comet assay
Cells seeded on 6-cm dishes (HCT116-p53+/+: 1 × 105 cells/well and CaCO-2: 1.5 × 105 cells/well) were treated with different LA concentrations and the respective solvent controls for 24 and 48 h. As positive control, the free radical generator t-BOOH (30 µM) was used. The cells were harvested and subjected to alkaline comet assay as described previously (Dörsam et al. 2014). Briefly, cells embedded in 0.5 % low melting point agarose were transferred onto an agarose-precoated slide and allowed to settle. Subsequently, the slides were incubated in lysis buffer consisting of 2.5 M NaCl, 100 mM EDTA, 1 % sodium lauroyl sarcosinate, 10 mM Tris, pH 10 for 1 h, which was followed by an unwinding step in electrophoresis buffer comprising 300 mM NaOH and 1 mM EDTA pH 13 for 25 min at 4 °C. Electrophoresis was then carried out for 15 min at 25 V and 300 mA, respectively, and samples were neutralized three times with 0.4 M Tris pH 7.5. After final fixation in 100 % ethanol, samples were allowed to air-dry and stained with 50 µg/mL propidium iodide. Obtained comets were analyzed by microscopy using an Olympus BX50 equipped with a ColorView camera (Olympus, Münster, Germany). In each experiment, 50 cells were scored using Comet IV software (Perceptive Instruments Ltd., Bury St Edmunds, UK).
Immunofluorescence-based analysis of γ-H2AX foci
Analysis of γ-H2AX foci was performed as described previously (Fahrer et al. 2014). Cells seeded on cover slips (HCT116-p53+/+: 1 × 105 cells/well and CaCO-2: 1 × 105 cells/well) were treated with 1 mM LA for 24 h or irradiated with 10 Gy using a Cs137 source. Cells were then fixed for 15 min in 4 % paraformaldehyde in PBS and in ice-cold methanol for 10 min at −20 °C. They were blocked in PBS containing 5 % BSA and 0.3 % Triton X-100 for 1 h at RT followed by incubation with a primary antibody against S139 phosphorylated histone H2AX for 1 h. After several washing steps, incubation with a secondary antibody coupled with Alexa Fluor 488 (Life Technologies, Darmstadt, Germany) was performed for 1 h. Finally, nuclei were counterstained with TO-PRO-3 diluted in PBS (Life Technologies, Darmstadt, Germany). Cells were then mounted using Vectashield® (Vector Labs, Burlingame, CA, USA) and analyzed by confocal microscopy with a Zeiss Axio Observer.Z1 microscope equipped with a LSM710 laser scanning unit (Zeiss, Oberkochen, Germany). Images were acquired in optical sections of 1 µm and processed with ImageJ (NIH, USA). Mean number of γ-H2AX foci per nucleus was assessed with ImageJ (at least 100 cells/treatment, n = 2), and data were evaluated by GraphPad Prism software.
SDS-PAGE and immunoblot analysis
Upon treatment, whole cell lysis was performed in buffer containing 25 mM Tris–HCl pH 8.0, 5 mM EDTA, 1 mM DTT, 0.5 M NaCl supplemented with complete protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). Protein content of clarified extracts was determined with the Bradford assay. Equal amounts of protein samples were separated by SDS-PAGE followed by transfer to a nitrocellulose membrane (Perkin Elmer, Rodgau, Germany) with a wet blot chamber (BioRad, München, Germany). Five percentage of (w/v) nonfat dry milk in TBS containing Tween-20 [0.1 % (v/v), TBS-T] was used to block the membrane for 1 h at room temperature (RT). Subsequently, the membrane was probed with the respective primary antibody diluted in TBS-T for 1 h at RT. After washing the membrane with TBS-T, it was incubated with the appropriate secondary antibody coupled to horseradish peroxidase (Santa Cruz Biotechnology, Heidelberg, Germany) for 1 h. After further washing steps, proteins were detected by chemiluminescence using Western Lightning® Plus-ECL (Perkin Elmer, Rodgau, Germany).
Reproduction of experiments and statistical analysis
All experiments were performed independently at least twice. Results from representative experiments are shown. Values (n ≥ 3) are presented as means ± standard errors of the means (SEM) using GraphPad Prism4 software. Statistical significance was calculated with Student’s t test.
Results
LA inhibits the growth of colorectal cancer cells and induces cell death in a p53-independent manner
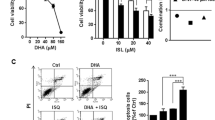
We first studied the effects of LA on the viability of CRC cells with different p53 status. HCT116 cells bearing wild-type p53 (Scian et al. 2008) as well as CaCO-2 and HT29 cells with mutant p53 (Kralj et al. 2003; Rodrigues et al. 1990) were treated with LA, and viability was determined using ATP assay. LA reduced cell viability in a dose-dependent manner in all CRC cell lines, with the strongest effect observed in HT29 cells (Fig. 1a–c). We then calculated the IC50 values for all cell lines tested, which ranged from 266 µM (HT29 cells) up to about 1,500 µM for HCT116 cells (Table 1 and supplementary information, Fig. S1). Owing to the higher cytotoxicity of LA detected in HT29 and CaCO-2 cells, we wished to address the role of p53 in LA-induced cytotoxicity in more detail. To this end, we used isogenic HCT116-p53+/+ and HCT116-p53−/− cells, which were challenged with increasing concentrations of LA for 72 h. Cell death was monitored by Annexin-V/PI staining and flow cytometry. LA-induced cell death increased dose-dependently and reached about 60 % at 2,000 µM LA in both cell lines (Fig. 2a). No effect was seen using ethanol as solvent control (Fig. 3h and data not shown). It should also be noted that only modest cell death rates were measured after 48 h of LA treatment (supplementary information, Fig. S2A). HCT116 cells incubated with 1,000 µM LA underwent striking morphological changes associated with cell death induction as compared to solvent-treated control cells (Fig. 2b). Subsequently, we determined the cell viability in both HCT116-p53+/+ and HCT116-p53−/− cells after LA treatment using the ATP assay. Almost no differences were observed between both isogenic cell lines, except for treatment with 2,000 µM LA that displayed a stronger effect in HCT116-p53 null cells (53 vs. 41 % in p53-wt cells). The IC50 value calculated for HCT116-p53−/− cells is very close to that of HCT116-p53+/+ cells (see Table 1; 1,584 µM vs. 1,514 µM), suggesting that p53 is dispensable for LA-mediated cytotoxicity. In addition, cells were exposed to the anticancer drugs doxorubicin (DR) and 5-FU. While no differences were determined in response to doxorubicin, HCT116-p53+/+ cells showed a significantly higher sensitivity toward 5-FU as compared to HCT116-p53−/− cells, which is in line with previous studies (Bunz et al. 1999).

Impact of LA on viability of colorectal cancer cells. a HCT116 cells (wt-p53) seeded in 96-well plates were treated with increasing LA concentrations as indicated. EtOH was included as solvent control. Cell viability was determined after 72 h by ATP assay and normalized to untreated control cells. b CaCO-2 and c HT29 cells were treated with LA as described in (a), and viability was determined with ATP assay. Data are presented as mean + SEM (n ≥ 3, triplicates). ns: not significant. ***p < 0.001 versus solvent control

LA induces p53-independent cell death in HCT116 cells. a HCT116-p53+/+ and HCT116-p53−/− cells were incubated with increasing LA doses for 72 h. Cell death was determined by Annexin-V/PI staining and flow cytometry. Data are presented as mean + SEM (n ≥ 3). **p < 0.005, ***p < 0.001 versus control. b Cells were treated with 1,000 µM LA or ethanol as solvent control for 72 h. Cell morphology was assessed by light microscopy with a 40 × oil immersion objective. Representative images are depicted, and the white bars correspond to 20 µm. c Measurement of cell viability in HCT116-p53+/+ and HCT116-p53−/− cells 72 h after LA treatment. The anticancer drugs doxorubicin (DR) and 5-fluorouracil (5-FU) were included as positive controls. Data are given as mean + SEM (n ≥ 3). **p < 0.005, ***p < 0.001. d Assessment of LA-induced loss of mitochondrial membrane potential. Both cell lines were treated with 1,000 µM LA or the respective negative controls. Valinomycin (VM) served as positive control. Following JC-1 staining, red and green fluorescence was measured by flow cytometry. Cells with depolarized mitochondria (green fluorescence) are shown. Data are presented as mean + SEM (n ≥ 3). **p < 0.005, ***p < 0.001 versus solvent control. e Cell cycle distribution in HCT116-p53+/+ cells following treatment with 1,000 µM LA for 24 h. 5-FU (10 µM) was used as phase-specific anticancer drug (n ≥ 3). f Cell cycle distribution after 48 h (n ≥ 3). g Cell cycle distribution after 72 h (n = 4)
In order to analyze cell death following LA more precisely, LA-induced changes in the mitochondrial membrane potential were assessed using the dye JC-1 that enters mitochondria in an electrochemical potential-dependent manner. LA caused a significant increase in the fraction of cells with compromised mitochondrial membrane potential, an early indicator of apoptosis, which was the same in both isogenic cell lines (Fig. 2d). As expected, the positive control valinomycin induced strong depolarization of mitochondria by permeabilization of the mitochondrial membrane for K+ ions. Finally, we tested whether LA is able to induce DNA fragmentation, which is a hallmark of apoptosis. Cells were treated for up to 72 h with LA and fixed followed by DNA staining with PI. Flow cytometry-based analysis showed LA-induced DNA fragmentation, as attested by a pronounced subG1 peak after 72 h, which was not observed in untreated control cells (Fig. 2f, g; supplementary information Fig. S2C and D). Cell cycle analysis also revealed a LA-induced accumulation of cells in G2/M-phase, which was already visible after 24 h and even more pronounced after 48 h as compared to control cells (Fig. 2e, f). This was accompanied by a reduction in the S-phase population and, to a less extent, the G1 population. It is interesting to note that the G2/M-block was relieved 72 h following LA treatment, in favor of a strongly increased subG1 population (Fig. 2g). The antimetabolite 5-FU (10 µM) was included as S-phase-specific anticancer drug. 5-FU provoked a clear G1 arrest after 24 h, which was followed by an increase in subG1 population after 48 h (Fig. 2e, f). A strong reduction in S-phase cells was observed 72 h after 5-FU treatment, coincident with G2/M arrest and a further increase in subG1 levels (Fig. 2g). DNA fragmentation upon LA treatment was corroborated by agarose gel electrophoresis, revealing LA-induced DNA laddering comparable to that of STS-treated cells (supplementary information, Fig. S2B). Taken together, LA reduced the viability of CRC cells in a dose-dependent manner. This was largely attributable to cell death induction, which obviously does not require wild-type p53.
LA induces caspase-dependent and caspase-independent cell death in HCT116 cells
Next, we dissected the mode of cell death induced by LA in HCT116 cells, which displayed Annexin-V positive cells and moderate loss of mitochondrial membrane potential indicative of apoptotic cell death (Fig. 2a, d). First, we monitored the involvement of caspases as key mediators of apoptosis (Pop and Salvesen 2009). Cells were incubated for 72 h with LA, and cleavage of the initiator caspases-9 was monitored by Western blot analysis. Caspase-9 is involved in the mitochondrial pathway of apoptosis, forming a complex with Apaf-1 and cytochrome C in the cytosol called apoptosome (Bao and Shi 2007). We observed a LA-dependent formation of a specific caspase-9 cleavage band, which was also detected in cells treated with the positive control STS for 8 h (Fig. 3a). However, we failed to detect a LA-dependent increase in caspase-9 activity, but rather observed an inhibition of caspase-9 in a dose-dependent manner (Fig. 3c). The positive control STS stimulated caspase-9 activity with a maximum 8 h after treatment (Fig. 3c), which is consistent with the observed STS-dependent peak in caspase-9 cleavage (Fig. 3a and supplementary information, Fig. S2E and F). We then assessed the cleavage and activation of the downstream caspases-3/7. Neither caspase-3 cleavage products nor an increase in caspase-3/7 activity could be detected following LA treatment, except for the positive control STS (Fig. 3b, d). Consistent with this finding, PARP-1 cleavage was also not observed following LA incubation, further arguing against the involvement of caspase-3/7 in LA-mediated cell death (Fig. 3b). In order to study a contribution of the extrinsic pathway of apoptosis, caspase-8 was detected by Western blot analysis. LA treatment did not result in the formation of caspase-8 cleavage fragments, whereas STS clearly induced caspase-8 cleavage. In addition, the impact of LA on caspase-2 was studied, which exhibits unique features of both initiator and executioner caspases (Vakifahmetoglu-Norberg and Zhivotovsky 2010) and is involved in the regulation of cell death triggered by metabolic imbalance and endoplasmic reticulum stress (Fava et al. 2012). Interestingly, we found an obvious LA-dependent degradation of full-length caspase-2, with the effect at higher LA doses comparable to that of the positive control STS (Fig. 3f). Densitometric evaluation of three independent experiments confirmed a significant, dose-dependent cleavage of caspase-2 in response to LA (supplementary information, Fig. S3C). Furthermore, the contribution of caspases to LA-induced death was investigated by the pan-caspase inhibitor zVAD. Co-treatment of cells with zVAD reduced LA-mediated cell demise independent of the p53 status in HCT116 cells (Fig. 3g and supplementary information, Fig. S3A and B). To test a possible involvement of poly(ADP-ribose) formation as a trigger of caspase-independent cell death (so-called Parthanatos), the potent PARP inhibitor Olaparib was used (Mangerich and Bürkle 2011), which, however, failed to block LA-induced cell death (supplementary information, Fig. S3D). We then wished to assess whether or not LA may stimulate necroptosis, a form of regulated caspase-independent cell death. Cells were treated with LA in combination with Necrostatin-1, which targets RIP1 kinase and thereby blocks necroptosis (Degterev et al. 2008). Interestingly, Necrostatin-1 co-treatment led to a significant decrease in LA-induced cell death (Fig. 3h), indicating that necroptosis contributes to caspase-independent cell death in HCT116 cells. In summary, our findings indicate caspase-dependent and caspase-independent mechanisms of cell demise in HCT116 cells following LA treatment.

LA-mediated cell death in HCT116 cells does not involve caspase-8 and caspase-3/7. HCT116-p53+/+ cells were exposed to increasing concentrations of LA for 72 h as indicated. Staurosporine (STS), a potent inducer of apoptosis, was included as positive control, and ethanol (EtOH) was used as solvent control. a Detection of caspase-9 cleavage. Samples were subjected to SDS-PAGE followed by detection with antibody against cleaved caspase-9. Hsp90 was visualized as loading control. b Assessment of caspase-3 and PARP-1 cleavage using immunoblot detection with an antibody against PARP-1 and its cleavage fragment as well as cleaved caspase-3. Hsp90 served as loading control. Representative immunoblots are shown. c Determination of caspase-9 activity using Caspase-Glo® 9 Assay. Data are given as mean + SEM (n = 3). ns: not significant, *p < 0.05, ***p < 0.001 versus solvent control. d Determination of caspase-3/7 activity using Caspase-Glo® 3/7 Assay. Data are given as mean + SEM (n = 3). ns: not significant, ***p < 0.001 versus solvent control. e Western blot analysis of caspase-8 and f caspase-2 cleavage. Representative blots are shown. g Cells were supplemented with the pan-caspase inhibitor zVAD (20 µM) after 24 and 48 h. Cell death was determined using flow cytometry following Annexin-V/PI staining. Light gray bars represent early apoptotic cells; dark gray bars depict late apoptotic/necrotic cell populations. Data are given as mean + SEM (n = 3). ns: not significant. h Cells were supplemented with 50 µM Necrostatin-1, a RIP1-kinase inhibitor that blocks necroptosis, 24 and 48 h after LA treatment. Cell death was measured as described above. Data are given as mean + SEM (n = 3). **p < 0.01
LA activates the intrinsic apoptosis pathway in CaCO-2 cells
To analyze cell death induction by LA, CaCO-2 cells were incubated with LA for 72 h and Annexin-V/PI staining was performed. LA caused a dose-dependent increase in the fraction of both early apoptotic and late apoptotic/necrotic cells (Fig. 4a). In line with these findings, cells displayed striking morphological changes with features of apoptosis (e.g., membrane blebbing and apoptotic bodies) after treatment with 750 µM LA, whereas cells incubated with the solvent control ethanol exhibited normal morphology and high confluency (Fig. 4b). In order to study whether LA stimulates the intrinsic apoptosis pathway, cells were subjected to Western blot analysis. Proteolytic degradation of caspase-9 occurred in a LA-dependent manner, which was already visible at 500 µM LA, whereas no effect was observed in solvent-treated control cells (Fig. 4c, top panel). The LA-induced degradation of caspase-9 coincided with the formation of its specific cleavage product (Fig. 4c, bottom panel). To confirm the activation of caspase-9, cells were treated for 72 h with LA and its activity was determined, revealing LA-dependent caspase-9 activation (Fig. 4e), which is consistent with its proteolytic degradation mentioned above (Fig. 4c). In order to assess a contribution of the extrinsic pathway of apoptosis, caspase-8 was monitored by Western blot analysis, but no caspase-8 cleavage fragments were visualized (supplementary information, Fig. S4A). Subsequently, the degradation of the executioner caspase-3 was analyzed and showed the formation of specific caspase-3 cleavage products in response to LA (Fig. 4d), which was corroborated by a pronounced dose-dependent increase in caspase-3/7 activity after 72 h (Fig. 4f). Solvent-treated control cells also exhibited a slight increase over untreated control cells, being, however, almost tenfold lower than the corresponding LA dose (750 µM). In agreement with the stimulation of caspase-3/7 activity, we were able to detect PARP-1 cleavage, a hallmark of apoptosis mediated by caspase-3/7 activity (Fig. 4d) (Soldani and Scovassi 2002). To assess the kinetics of cell death induction by LA, we further analyzed caspase-3/7 activation and PARP-1 cleavage already after 48 h of LA treatment. We did not observe an increase in caspase-3/7 activity and detected only very little PARP-1 cleavage after 48 h (supplementary information, Fig. S4B and C), suggesting that LA induces cell death primarily after incubation over longer time periods, i.e., 72 h. We then used the pan-caspase inhibitor zVAD to block LA-induced cell death. Supplementation of LA-treated cells with zVAD resulted in a pronounced decrease in cell death, however, without reaching statistical significance (Fig. 4g). Immunoblot detection revealed lack of caspase-9 cleavage and impaired PARP-1 and caspase-3 cleavage in cells co-treated with LA and zVAD, which is in agreement with the reduced cell death induction (Fig. 4h). In addition, experiments were also conducted with HT29 cells, revealing similar caspase-dependent cell death (supplementary information, Fig. S5). Collectively, the results show that LA induces caspase-dependent cell death in CaCO-2 cells, involving the activation of caspase-9 and caspase-3/7.

LA triggers caspase-dependent cell death in CaCO-2 cells. a Cells were treated with LA and processed for Annexin-V/PI staining. Data are presented as mean + SEM (n = 3). ns: not significant. **p < 0.005, ***p < 0.001 versus solvent control. b Cells were treated with 750 µM LA or ethanol as solvent control for 72 h. Cell morphology was monitored by light microscopy using a 40 × oil immersion objective. Representative images are shown, and white bars correspond to 20 µm. c Cells were treated with increasing concentrations of LA as indicated. EtOH was used as solvent control. Cells were harvested after 72 h, and samples were separated by SDS-PAGE followed by immunoblot detection with a caspase-9 and cleaved caspase-9 antibody, respectively. Hsp90 was visualized as loading control. d Detection of caspase-3 and PARP-1 cleavage in cells processed as mentioned above. Hsp90 served as loading control. Representative Western blots are depicted. e Assessment of caspase-9 activity using a luminogenic substrate 72 h after incubation with LA. Data are expressed as mean + SEM (n = 3). ns: not significant. ***p < 0.001 versus solvent control. f Assessment of caspase-3/7 activity using a luminogenic substrate 72 h after incubation with LA. Data are expressed as mean + SEM (n = 3). ns: not significant. *p < 0.05, ***p < 0.001 versus solvent control. G Cells were supplemented with the pan-caspase inhibitor zVAD 24 and 48 h after LA treatment. Cell death was assessed by Annexin-V/PI staining and flow cytometry. Data are expressed as mean + SEM (n = 3). ns: not significant. h Cells were treated with LA and zVAD as described. Samples were subjected to SDS-PAGE followed by Western blot analysis for cleaved caspase-3 and caspase-9 as well as PARP-1. Representative blots are shown
LA activates p21 in a p53-independent manner and does not exhibit genotoxicity in CRC cells
p53 is a central player in the maintenance of genomic integrity and is activated by the DNA damage response, resulting in cell cycle arrest and/or apoptosis (Horn and Vousden 2007). To analyze a potential activation or stabilization of p53 by LA, HCT116-p53+/+ cells were treated with LA over 24 h. No increase in p53 protein level was detected, whereas the antineoplastic agent doxorubicin effectively induced p53 accumulation (Fig. 5a). LA did also not stabilize p53 at earlier time points nor did a lower LA dose (100 µM) have any impact on p53 level (supplementary information, Fig. S6A and B). However, LA treatment led to a strong induction of p21, a known downstream target of p53 (Fig. 5a, bottom panel). Strikingly, a comparable upregulation of p21 was also found in HCT116-p53−/− cells, suggesting a p53-independent mechanism triggered by LA. The lack of p53 activation in response to LA is consistent with the finding that LA-induced cytotoxicity is independent of p53 (see above). It has been reported previously that LA interferes with the cellular redox balance in cancer cells by either promoting ROS formation (Michikoshi et al. 2013; Moungjaroen et al. 2006) or decreasing ROS levels (Dozio et al. 2010; Shi et al. 2008). To determine the effects of LA on endogenous ROS levels, HCT116 were incubated for 24 h with a low (200 µM) or a high (2,000 µM) dose of LA and stained with the redox-sensitive probe CM-H2DCFDA followed by flow cytometry. 200 µM LA showed an antioxidative effect with a decrease in cellular ROS levels as compared to solvent-treated control cells (Fig. 5b). The high doses of LA elicited a slight, but not significant induction of ROS levels, whereas the positive control H2O2 massively enhanced cellular ROS. CaCO-2 cells were also challenged with LA for 24 h, showing comparable effects (supplementary information, Fig. S6C). Next, we monitored the phosphorylation of histone 2AX (γ-H2AX), a well-established surrogate marker for DNA double-strand breaks (DSBs) (Kinner et al. 2008). Confocal microscopy revealed no induction of γ-H2AX following treatment with 1,000 µM LA for 24 h (Fig. 5c, middle panel). As positive control, cells were irradiated with 10 Gy and incubated for 30 min, which resulted in the formation of numerous discrete γ-H2AX foci (Fig. 5c, bottom panel). Quantitative evaluation confirmed that LA did not increase the γ-H2AX level beyond that of untreated control cells. In contrast to HCT116 cells, CaCO-2 cells displayed a higher number of γ-H2AX foci in untreated controls and a small, but significant increase in LA-treated cells (Fig. 5d). These findings were corroborated by immunoblot detection of γ-H2AX (supplementary information, Fig. S6D and E).

DNA damage response and genotoxicity of LA in CRC cells. a Impact of LA on p53 and p21 in HCT116 cells. HCT116-p53+/+ and HCT116-p53−/− cells were incubated with 1,000 µM LA for 24 h. Ethanol was used as solvent control, and the DNA-damaging agent doxorubicin (4 µM) was included as positive control. Cells lysates were subjected to SDS-PAGE and immunoblot analysis for p53, p21 and Hsp90 (loading control). b Determination of ROS levels in HCT116 cells. Cells were treated with LA for 24 h. In addition, H2O2 was included as positive control. After washing, cells were incubated with the ROS sensor CM-H2DCFDA, and ROS level was determined by flow cytometry. Data are expressed as mean + SEM (n = 3). ns: not significant. **p < 0.005 versus solvent control. c HCT116 cells were treated with LA or the corresponding solvent control. As positive control, cells were irradiated with 10 Gy. After fixation, cells were stained with a γ-H2AX antibody followed by an Alexa 488-coupled secondary antibody (green). Nuclei were counterstained with TO-PRO-3 (blue). Images were acquired by confocal microscopy and processed by ImageJ software. Representative images are shown. D Quantitative evaluation of γ-H2AX in HCT116 and CaCO-2 cells. Number of γ-H2AX foci per nucleus was determined by ImageJ software and further evaluated with GraphPad Prism 5.0 (50 - 100 cells per experiment; n = 2). Data are expressed as mean + SEM. ns: not significant. *p < 0.05, ***p < 0.001 versus control. E Assessment of DNA damage by alkaline comet assay after treatment of HCT116 and CaCO-2 cells for 24 h with LA. t-BOOH was used as positive control. At least 50 cells per experiment were analyzed using comet IV software. Results displayed are the mean + SEM (n = 2). ns: not significant. *p < 0.05, ***p < 0.001 versus solvent control (color figure online)
In order to screen for other forms of DNA damage, an alkaline comet assay was performed that detects alkali-labile sites, DNA single-strand breaks (SSBs) and DSBs. HCT116 and CaCO-2 cells were incubated for 24 h with 1,000 or 2,000 µM LA and subjected to the assay. LA did not induce DNA damage in both cell lines, whereas the positive control t-BOOH caused high levels of DNA strand breaks (Fig. 5e). It should be noted that LA was also not positive in the alkaline comet assay at concentrations of 1,000 and 2,000 µM tested after 48 h (supplementary information, Fig. S6F). Taken together, we conclude that LA in the cytotoxic dose range bears no genotoxic potential as attested by lack of p53 stabilization and DNA damage induction in CRC cells.
LA potentiates 5-FU-induced cell death in HCT116 and CaCO-2 cells
Having demonstrated the cytotoxic activity of LA on CRC cells, we set out to characterize a putative synergy between LA and 5-FU, a classical anticancer drug used as “gold standard” in CRC therapy (Cunningham et al. 2010). HCT116 cells were treated with 5-FU or a combination of 5-FU and LA for 72 h, and cell viability was assessed by ATP assay. 5-FU led to a decrease in cell viability below 50 %, which was potentiated by concomitant LA treatment, reaching 25 % viability at a dose of 1,000 µM LA (Fig. 6a). To analyze a synergistic effect on cell death induction, cells were treated as above and subG1 population was assessed. 5-FU on its own caused a subG1 population of about 25 %, whereas addition of 500 µM LA increased the level to 34 % (Fig. 6b). Strikingly, 1,000 µM LA further enhanced 5-FU triggered cell death as attested by a subG1 population amounting to 39 %. These findings were corroborated by Annexin-V/PI staining, demonstrating a significant LA-dependent potentiation of 5-FU-induced cell death (Fig. 6c, d). Addition of 1,000 µM LA almost doubled the cell death rate in comparison with 5-FU single treatment (38 vs. 20 %).

Potentiation of 5-FU-induced cell death by LA in HCT116 cells. a Cells were treated with 5-FU, LA or a combination of both with LA as indicated. Cell viability was monitored by ATP assay after 72 h. Data are expressed as mean + SEM (n ≥ 2, triplicates). **p < 0.01, ***p < 0.001. b Cells were treated as described above, and subG1 population was determined by flow cytometry. Data are displayed as mean + SEM (n = 4). *p < 0.05, **p < 0.005, ***p < 0.001. c Cells were incubated as described above, stained with Annexin-V/PI and then analyzed by flow cytometry. Quantitative evaluation was performed by FACSDiva software. Data are given as mean + SEM (n = 2, triplicates). **p < 0.01, ***p < 0.001. d Representative dot plots are shown
CaCO-2 cells were also exposed to a combination of 5-FU and LA to confirm the synergistic cell killing in p53-mutated cells. Treatment with 5-FU or 5-FU together with LA showed a significant decrease in cell viability using the combination regime as compared to 5-FU single treatment (Fig. 7a). This effect was already significant at 200 µM LA and further enhanced at 500 µM LA. To reveal a synergistic effect on cell death induction, subG1 measurement was performed by flow cytometry, showing a significant enhancement of subG1 levels in cells treated with 5-FU and 1,000 µM LA as compared to 5-FU single treatment (Fig. 7b). However, no increase was observed as compared to LA single treatment. Finally, cell death induction was assessed by Annexin-V/PI staining. 5-FU administration caused a cell death rate of approximately 13 %, which was increased to 23 and 36 % at 500 and 1,000 µM LA, respectively (Fig. 7c, d). This level corresponds to that detected after LA single treatment, indicating that the cell death induction was largely attributable to LA. Collectively, our findings demonstrate that LA potentiates the cytotoxicity of 5-FU in CRC cells.

Potentiation of 5-FU-induced cell death by LA in CaCO-2 cells. a Cells were challenged with 5-FU, LA or a combination of both as indicated. 72 h later, cell viability was assessed by ATP assay. Data are expressed as mean + SEM (n ≥ 2, triplicates). *p < 0.05, **p < 0.01, ***p < 0.001. b Cells were treated as mentioned before, and subG1 population was determined by PI staining followed by flow cytometry. Data are displayed as mean + SEM (n = 2, duplicates). ns: not significant, *p < 0.05. c Cells were treated as described and subjected to Annexin-V/PI staining, which was analyzed by flow cytometry. Quantitative evaluation of Annexin-V/PI staining was conducted using FACSDiva software. Data are given as mean + SEM (n = 2, duplicates). **p < 0.01, ***p < 0.0001. d Representative dot plots are depicted
Discussion
In the present study, we analyzed the effect of LA, a naturally occurring and dithiol-containing antioxidant, on colorectal cancer cells and demonstrated a dose-dependent growth inhibition. The differences in LA sensitivity may relate to different expression levels of monocarboxylate transporter, which is important for its cell uptake (Takaishi et al. 2007) and/or divergent intracellular metabolism, e.g., conversion into DHLA (Handelman et al. 1994). Moreover, LA treatment may also trigger survival mechanisms in a cell line-specific manner that could account for lower cytotoxicity. In this regard, it has recently been described that a new synthetic LA derivative induced autophagy in HT29 cells (Hiratsuka et al. 2013), which is known to promote cancer cell survival (White 2012). Our results obtained from isogenic HCT116 cells differing in their p53 status further demonstrate that p53 is dispensable for LA-induced growth inhibition and cell death. The notion that LA acts in a p53-independent manner is supported by lack of p53 stabilization in HCT116 cells, and the higher cytotoxicity was observed in p53-deficient CaCO-2 and HT29 cells. These findings are in contrast to an earlier study, which indicated a higher sensitivity to LA in p53-proficient HCT116 cells as revealed by TUNEL staining (Yoo et al. 2013). Growth inhibition following LA was largely attributable to cell death induction that involved different cell death pathways.
In CaCO-2 and HT29 cells, LA induced the intrinsic apoptosis pathway characterized by activation of caspase-9 and caspase-3/7 with subsequent PARP-1 cleavage. This has also been reported for H460 lung cancer cells and HepG2 hepatoma cells, in which LA induced apoptosis via cytochrome C release and caspase-9 activation (Moungjaroen et al. 2006; Simbula et al. 2007). We were not able to detect caspase-8 cleavage in CaCO-2 cells, which is activated by extrinsic apoptotic stimuli such as Fas ligand/CD95 via Fas receptor (FasR) and Fas-associated protein with death domain (FADD) (Fulda and Debatin 2006). In line with this observation, a previous study demonstrated that cells deficient in FasR or FADD are not compromised during LA-induced apoptosis (van de Mark et al. 2003).
In HCT116 cells, LA treatment provoked caspase-9 cleavage as attested by Western blot analysis, which, however, was not associated with an increase in caspase-9 activity. Consistent with the lack of caspase-9 activity, the downstream effector caspase-3 was not stimulated following LA treatment and cleavage of its substrate PARP-1 was not detected. Nevertheless, HCT116 cells displayed several features of apoptosis including cell surface exposure of phosphatidylserine units and DNA fragmentation following LA exposure. In addition, treatment of HCT116 cells with the pan-caspase inhibitor zVAD suppressed LA-induced cell death, supporting the notion that caspases contribute to LA-induced cell death (but very likely not caspase-3, caspase-8 and caspase-9). Hence, we analyzed the putative cleavage of caspase-2 in response to LA treatment, which was shown to occur in a dose-dependent manner in HCT116 cells. Caspase-2 is unique in its ability to function as both initiator and effector caspase and is associated with cell death triggered by metabolic imbalance as well as ER stress (Fava et al. 2012). Interestingly, LA interferes with mitochondrial energy metabolism as pointed out below. Furthermore, LA might also induce ER stress, which was reported previously for the structurally related dithiol compound DTT (Hiramatsu et al. 2006). The observed dose-dependent cleavage of caspase-2 in response to LA treatment points to an involvement of caspase-2 in LA-mediated cell death and may explain the zVAD-dependent inhibition of cell death in HCT116 cells. Further studies with the inhibitor Necrostatin-1 provided evidence that caspase-independent mechanisms also contribute to LA-dependent cell death. Caspase-independent cell demise was also described in H460 and SaOS cells following treatment with CP-631, a derivative synthesized from LA (Zachar et al. 2011). However, the precise course of cell death in HCT116 cells remains to be determined and may also involve the release of toxic proteins such as AIF (Gogvadze et al. 2009) as a result of mitochondrial outer membrane permeabilization.
Next, we analyzed whether LA activates p53 and may bear genotoxic potential by damaging DNA directly or indirectly, e.g., by catalyzing ROS formation. Importantly, LA treatment did not result in p53 stabilization within 24 h, but rather decreased p53 protein level at a high dose of 1 mM. The LA-induced depletion of p53 protein was even more pronounced after 48 h and was dose-dependent (Göder et al., in preparation). This is in contrast to a previous study, which reported a LA-mediated stabilization of p53 (Yoo et al. 2013). Moreover, LA strongly induced the cell cycle regulator p21, which occurred in a p53-independent manner as demonstrated in isogenic HCT116-p53−/− cells. p21 expression is also controlled by p53-independent pathways and involves transcription factors such as STATs and CCAAT/enhancer-binding protein-β (C/EBP-β) (Abbas and Dutta 2009). Intriguingly, a p53-independent induction of p21 has already been shown for the thiol-containing antioxidant pyrrolidinedithiocarbamate (Chinery et al. 1997). Moreover, the increased p21 levels following LA treatment are associated with an accumulation of HCT116 cells in G2/M-phase, which is already detectable after 24 h and even more pronounced after 48 h. This is consistent with the notion that p21 blocks the kinase activity of CDK1, resulting in a cell cycle arrest in G2-phase (Abbas and Dutta 2009). No significant elevation of cellular ROS levels was observed at high LA doses, while lower doses displayed a pronounced antioxidative effect. This is in agreement with the notion that LA is a potent inhibitor of ROS generation and has also been described in other tumor cell lines challenged with LA (Dozio et al. 2010; Shi et al. 2008). Induction of DNA strand breaks was measured by the alkaline comet assay and revealed no genotoxic effect of LA. This lack of DNA strand breaks is consistent with the level of γ-H2AX that is unaffected following LA treatment in HCT116 cells. A slight induction of γ-H2AX was detectable in CaCO-2 cells, which could also result from LA-mediated PP2A inhibition. PP2A is a protein phosphatase that is responsible for the dephosphorylation of γ-H2AX (Chowdhury et al. 2005) and was shown to be partially inhibited by LA (Shay and Hagen 2009).
Finally, we analyzed whether or not LA potentiates the cytotoxic effects of 5-FU on colorectal cancer cells. 5-FU is an anticancer drug that is used as first-line treatment in adjuvant chemotherapy of CRC (Cunningham et al. 2010). 5-FU and its metabolites act primarily on DNA and RNA, resulting in cytotoxicity (Pettersen et al. 2011). Strikingly, a combination based on 5-FU and LA reduced cell viability and significantly potentiated cell death in HCT116 and CaCO-2 cells. Both cell lines completely differ in their p53 status, which implies that the observed effects are independent of functional p53. This is an important aspect since p53 is inactivated by mutations in the majority of CRC cell lines (Liu and Bodmer 2006) and in about 50 % of all human cancers (Olivier et al. 2010). CRC cells deficient in p53 are also more resistant to 5-FU (Bunz et al. 1999), which may be overcome by supplementation with LA due to its p53-independent mode of action.
Derived from octanoic acid, LA shares structural homology with short fatty acids such as butyrate and propionate, which are known to induce apoptosis in CRC cells in the millimolar range (Marques et al. 2013; Ruemmele et al. 2003). We showed that already lower concentrations of LA (250–500 µM) are sufficient to efficiently trigger cell death in different CRC cell lines. This concentration range is comparable to other reports dealing with the cytotoxicity of LA on tumor cells (Dozio et al. 2010; Michikoshi et al. 2013; Shi et al. 2008; Simbula et al. 2007). Different routes of administration have been described for humans, including oral administration and intravenous (i.v.) injection. Oral supplementation with 600 mg LA resulted in maximal plasma concentration (C max) of up to 150 µM, while i.v. injection of 300 mg racemic LA yielded a C max of 110 µM (Carlson et al. 2007). Interestingly, LA has been given at doses up to 1,200 mg intravenously and up to 2,400 mg orally without observing severe side effects (Reljanovic et al. 1999; Yadav et al. 2005; Ziegler et al. 1995). Based on these doses, C max values of up to 500 µM can be estimated, which is at or above the IC50 values determined for the p53-mutated CRC cell lines in the present study. Due to its non-genotoxic properties shown here and preferential toxicity on tumor cells described before (Pack et al. 2002; van de Mark et al. 2003; Wenzel et al. 2005), LA might offer several advantages such as reduced adverse effects in healthy tissue as compared to classical anticancer drugs. It may even counteract colorectal carcinogenesis in early stages due to its anti-inflammatory action in the gut (Trivedi and Jena 2013).
LA is tightly linked to mitochondrial energy metabolism owing to its vital function as cofactor of pyruvate dehydrogenase (PDH) (Shay et al. 2009). It is well established that cancer cells display an altered energy metabolism characterized by higher glucose uptake and generation of lactate independent of the oxygen level, conferring a proliferative advantage (Vander Heiden et al. 2009). This type of aerobe glycolysis is referred to as “Warburg effect” and is an enabling hallmark of cancer cells (Hanahan and Weinberg 2011). PDH is the central player in this network and, in concert with lactate dehydrogenase, governs the switch between glycolysis and oxidative phosphorylation. PDH activity is negatively regulated by pyruvate dehydrogenase kinase (PDK) by inhibitory phosphorylation, which is overexpressed in cancers and induced by hypoxia-inducible factor-1 (HIF-1) (Kim et al. 2006). Pharmacological inhibition of PDK restores PDH activity and suppresses tumor growth in vivo (Bonnet et al. 2007). Interestingly, LA was also shown to block PDK and decreased the level of PDH phosphorylation, which in turn stimulates PDH activity (Korotchkina et al. 2004). It is therefore tempting to speculate that LA-mediated restoration of PDH activity may contribute to its cytotoxicity observed in CRC cells and other tumor cell lines as described herein and elsewhere (Dozio et al. 2010; Michikoshi et al. 2013; Moungjaroen et al. 2006; Pack et al. 2002; Shi et al. 2008; van de Mark et al. 2003).
Collectively, our findings demonstrate that LA induces CRC cell death independent of their p53 status and increases the 5-FU-mediated cytotoxicity without causing DNA damage on its own. The application of LA together with 5-FU may provide a novel strategy for targeting CRC cells in cancer therapy.
References
Abbas T, Dutta A (2009) p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 9(6):400–414. doi:10.1038/nrc2657
Bao Q, Shi Y (2007) Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ 14(1):56–65. doi:10.1038/sj.cdd.4402028
Bonnet S, Archer SL, Allalunis-Turner J et al (2007) A mitochondria-K + channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11(1):37–51. doi:10.1016/j.ccr.2006.10.020
Bunz F, Hwang PM, Torrance C et al (1999) Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Investig 104(3):263–269. doi:10.1172/JCI6863
Carlson DA, Smith AR, Fischer SJ, Young KL, Packer L (2007) The plasma pharmacokinetics of R-(+)-lipoic acid administered as sodium R-(+)-lipoate to healthy human subjects. Altern Med Rev 12(4):343–351
Chinery R, Brockman JA, Peeler MO, Shyr Y, Beauchamp RD, Coffey RJ (1997) Antioxidants enhance the cytotoxicity of chemotherapeutic agents in colorectal cancer: a p53-independent induction of p21WAF1/CIP1 via C/EBPbeta. Nat Med 3(11):1233–1241
Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J (2005) gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell 20(5):801–809. doi:10.1016/j.molcel.2005.10.003
Cunningham D, Atkin W, Lenz HJ et al (2010) Colorectal cancer. Lancet 375(9719):1030–1047. doi:10.1016/S0140-6736(10)60353-4
Degterev A, Hitomi J, Germscheid M et al (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4(5):313–321. doi:10.1038/nchembio.83
Dörsam B, Wu CF, Efferth T, Kaina B, Fahrer J (2014) The eucalyptus oil ingredient 1,8-cineol induces oxidative DNA damage. Arch Toxicol. doi:10.1007/s00204-014-1281-z
Dozio E, Ruscica M, Passafaro L et al (2010) The natural antioxidant alpha-lipoic acid induces p27(Kip1)-dependent cell cycle arrest and apoptosis in MCF-7 human breast cancer cells. Eur J Pharmacol 641(1):29–34. doi:10.1016/j.ejphar.2010.05.009
Fahrer J, Kaina B (2013) O6-methylguanine-DNA methyltransferase in the defense against N-nitroso compounds and colorectal cancer. Carcinogenesis 34(11):2435–2442. doi:10.1093/carcin/bgt275
Fahrer J, Huelsenbeck J, Jaurich H et al (2014) Cytolethal distending toxin (CDT) is a radiomimetic agent and induces persistent levels of DNA double-strand breaks in human fibroblasts. DNA Repair 18:31–43. doi:10.1016/j.dnarep.2014.03.002
Fava LL, Bock FJ, Geley S, Villunger A (2012) Caspase-2 at a glance. J Cell Sci 125(Pt 24):5911–5915. doi:10.1242/jcs.115105
Fulda S, Debatin KM (2006) Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25(34):4798–4811. doi:10.1038/sj.onc.1209608
Gogvadze V, Orrenius S, Zhivotovsky B (2009) Mitochondria as targets for cancer chemotherapy. Semin Cancer Biol 19(1):57–66. doi:10.1016/j.semcancer.2008.11.007
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. doi:10.1016/j.cell.2011.02.013
Handelman GJ, Han D, Tritschler H, Packer L (1994) Alpha-lipoic acid reduction by mammalian cells to the dithiol form, and release into the culture medium. Biochem Pharmacol 47(10):1725–1730
Hiramatsu N, Kasai A, Hayakawa K, Yao J, Kitamura M (2006) Real-time detection and continuous monitoring of ER stress in vitro and in vivo by ES-TRAP: evidence for systemic, transient ER stress during endotoxemia. Nucleic Acids Res 34(13):e93. doi:10.1093/nar/gkl515
Hiratsuka T, Inomata M, Kono Y, Yokoyama S, Shiraishi N, Kitano S (2013) DHLTauZnNa, a newly synthesized alpha-lipoic acid derivative, induces autophagy in human colorectal cancer cells. Oncol Rep 29(6):2140–2146. doi:10.3892/or.2013.2394
Horn HF, Vousden KH (2007) Coping with stress: multiple ways to activate p53. Oncogene 26(9):1306–1316. doi:10.1038/sj.onc.1210263
Jones W, Li X, Qu ZC, Perriott L, Whitesell RR, May JM (2002) Uptake, recycling, and antioxidant actions of alpha-lipoic acid in endothelial cells. Free Radic Biol Med 33(1):83–93
Kim JW, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3(3):177–185. doi:10.1016/j.cmet.2006.02.002
Kinner A, Wu W, Staudt C, Iliakis G (2008) Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res 36(17):5678–5694. doi:10.1093/nar/gkn550
Kopetz S, Chang GJ, Overman MJ et al (2009) Improved survival in metastatic colorectal cancer is associated with adoption of hepatic resection and improved chemotherapy. J Clin Oncol 27(22):3677–3683. doi:10.1200/JCO.2008.20.5278
Korotchkina LG, Sidhu S, Patel MS (2004) R-lipoic acid inhibits mammalian pyruvate dehydrogenase kinase. Free Radic Res 38(10):1083–1092. doi:10.1080/10715760400004168
Kralj M, Husnjak K, Korbler T, Pavelic J (2003) Endogenous p21WAF1/CIP1 status predicts the response of human tumor cells to wild-type p53 and p21WAF1/CIP1 overexpression. Cancer Gene Ther 10(6):457–467. doi:10.1038/sj.cgt.7700588
Liu Y, Bodmer WF (2006) Analysis of P53 mutations and their expression in 56 colorectal cancer cell lines. Proc Natl Acad Sci USA 103(4):976–981. doi:10.1073/pnas.0510146103
Mangerich A, Bürkle A (2011) How to kill tumor cells with inhibitors of poly(ADP-ribosyl)ation. Int J Cancer 128(2):251–265. doi:10.1002/ijc.25683
Marques C, Oliveira CS, Alves S et al (2013) Acetate-induced apoptosis in colorectal carcinoma cells involves lysosomal membrane permeabilization and cathepsin D release. Cell Death Dis 4:e507. doi:10.1038/cddis.2013.29
Michikoshi H, Nakamura T, Sakai K et al (2013) Alpha-lipoic acid-induced inhibition of proliferation and met phosphorylation in human non-small cell lung cancer cells. Cancer Lett 335(2):472–478. doi:10.1016/j.canlet.2013.03.008
Moungjaroen J, Nimmannit U, Callery PS et al (2006) Reactive oxygen species mediate caspase activation and apoptosis induced by lipoic acid in human lung epithelial cancer cells through Bcl-2 down-regulation. J Pharmacol Exp Ther 319(3):1062–1069. doi:10.1124/jpet.106.110965
Olivier M, Hollstein M, Hainaut P (2010) TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2(1):a001008. doi:10.1101/cshperspect.a001008
Pack RA, Hardy K, Madigan MC, Hunt NH (2002) Differential effects of the antioxidant alpha-lipoic acid on the proliferation of mitogen-stimulated peripheral blood lymphocytes and leukaemic T cells. Mol Immunol 38(10):733–745
Pettersen HS, Visnes T, Vagbo CB et al (2011) UNG-initiated base excision repair is the major repair route for 5-fluorouracil in DNA, but 5-fluorouracil cytotoxicity depends mainly on RNA incorporation. Nucleic Acids Res 39(19):8430–8444. doi:10.1093/nar/gkr563
Pop C, Salvesen GS (2009) Human caspases: activation, specificity, and regulation. J Biol Chem 284(33):21777–21781. doi:10.1074/jbc.R800084200
Prasad PD, Wang H, Kekuda R et al (1998) Cloning and functional expression of a cDNA encoding a mammalian sodium-dependent vitamin transporter mediating the uptake of pantothenate, biotin, and lipoate. J Biol Chem 273(13):7501–7506
Reljanovic M, Reichel G, Rett K et al (1999) Treatment of diabetic polyneuropathy with the antioxidant thioctic acid (alpha-lipoic acid): a two year multicenter randomized double-blind placebo-controlled trial (ALADIN II). Alpha lipoic acid in diabetic neuropathy. Free Radic Res 31(3):171–179
Rochette L, Ghibu S, Richard C, Zeller M, Cottin Y, Vergely C (2013) Direct and indirect antioxidant properties of alpha-lipoic acid and therapeutic potential. Mol Nutr Food Res 57(1):114–125. doi:10.1002/mnfr.201200608
Rodrigues NR, Rowan A, Smith ME et al (1990) p53 mutations in colorectal cancer. Proc Natl Acad Sci USA 87(19):7555–7559
Ruemmele FM, Schwartz S, Seidman EG, Dionne S, Levy E, Lentze MJ (2003) Butyrate induced Caco-2 cell apoptosis is mediated via the mitochondrial pathway. Gut 52(1):94–100
Scian MJ, Carchman EH, Mohanraj L et al (2008) Wild-type p53 and p73 negatively regulate expression of proliferation related genes. Oncogene 27(18):2583–2593. doi:10.1038/sj.onc.1210898
Shay KP, Hagen TM (2009) Age-associated impairment of Akt phosphorylation in primary rat hepatocytes is remediated by alpha-lipoic acid through PI3 kinase, PTEN, and PP2A. Biogerontology 10(4):443–456. doi:10.1007/s10522-008-9187-x
Shay KP, Moreau RF, Smith EJ, Smith AR, Hagen TM (2009) Alpha-lipoic acid as a dietary supplement: molecular mechanisms and therapeutic potential. Biochim Biophys Acta 1790(10):1149–1160. doi:10.1016/j.bbagen.2009.07.026
Shi DY, Liu HL, Stern JS, Yu PZ, Liu SL (2008) Alpha-lipoic acid induces apoptosis in hepatoma cells via the PTEN/Akt pathway. FEBS Lett 582(12):1667–1671. doi:10.1016/j.febslet.2008.04.021
Simbula G, Columbano A, Ledda-Columbano GM et al (2007) Increased ROS generation and p53 activation in alpha-lipoic acid-induced apoptosis of hepatoma cells. Apoptosis 12(1):113–123. doi:10.1007/s10495-006-0487-9
Smith AR, Shenvi SV, Widlansky M, Suh JH, Hagen TM (2004) Lipoic acid as a potential therapy for chronic diseases associated with oxidative stress. Curr Med Chem 11(9):1135–1146
Soldani C, Scovassi AI (2002) Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis Int J Progr Cell Death 7(4):321–328
Suh JH, Shenvi SV, Dixon BM et al (2004) Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci USA 101(10):3381–3386. doi:10.1073/pnas.0400282101
Takaishi N, Yoshida K, Satsu H, Shimizu M (2007) Transepithelial transport of alpha-lipoic acid across human intestinal Caco-2 cell monolayers. J Agric Food Chem 55(13):5253–5259. doi:10.1021/jf063624i
Teichert J, Kern J, Tritschler HJ, Ulrich H, Preiss R (1998) Investigations on the pharmacokinetics of alpha-lipoic acid in healthy volunteers. Int J Clin Pharmacol Ther 36(12):625–628
Trivedi PP, Jena GB (2013) Role of alpha-lipoic acid in dextran sulfate sodium-induced ulcerative colitis in mice: studies on inflammation, oxidative stress, DNA damage and fibrosis. Food Chem Toxicol 59:339–355. doi:10.1016/j.fct.2013.06.019
Vakifahmetoglu-Norberg H, Zhivotovsky B (2010) The unpredictable caspase-2: what can it do? Trends Cell Biol 20(3):150–159. doi:10.1016/j.tcb.2009.12.006
van de Mark K, Chen JS, Steliou K, Perrine SP, Faller DV (2003) Alpha-lipoic acid induces p27Kip-dependent cell cycle arrest in non-transformed cell lines and apoptosis in tumor cell lines. J Cell Physiol 194(3):325–340. doi:10.1002/jcp.10205
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324(5930):1029–1033. doi:10.1126/science.1160809
Wenzel U, Nickel A, Daniel H (2005) Alpha-lipoic acid induces apoptosis in human colon cancer cells by increasing mitochondrial respiration with a concomitant O2-*-generation. Apoptosis 10(2):359–368. doi:10.1007/s10495-005-0810-x
White E (2012) Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 12(6):401–410. doi:10.1038/nrc3262
Yadav V, Marracci G, Lovera J et al (2005) Lipoic acid in multiple sclerosis: a pilot study. Multiple sclerosis 11(2):159–165
Yoo TH, Lee JH, Chun HS, Chi SG (2013) alpha-Lipoic acid prevents p53 degradation in colon cancer cells by blocking NF-kappaB induction of RPS6KA4. Anticancer Drugs 24(6):555–565. doi:10.1097/CAD.0b013e32836181eb
Zachar Z, Marecek J, Maturo C et al (2011) Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J Mol Med (Berl) 89(11):1137–1148. doi:10.1007/s00109-011-0785-8
Ziegler D, Hanefeld M, Ruhnau KJ et al (1995) Treatment of symptomatic diabetic peripheral neuropathy with the anti-oxidant alpha-lipoic acid. A 3-week multicentre randomized controlled trial (ALADIN Study). Diabetologia 38(12):1425–1433
Acknowledgments
The author would like to thank Dr. Bert Vogelstein (Baltimore, USA) for providing HCT116 cells. We are grateful to Anna Frumkina (Mainz, Germany) for performing the alkaline comet assay. This work was supported by the University of Mainz (Stufe I, grant to J.F.) and University Medical Center Mainz (MAIFOR).
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dörsam, B., Göder, A., Seiwert, N. et al. Lipoic acid induces p53-independent cell death in colorectal cancer cells and potentiates the cytotoxicity of 5-fluorouracil. Arch Toxicol 89, 1829–1846 (2015). https://doi.org/10.1007/s00204-014-1434-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-014-1434-0