
Abstract
A Gram-stain-negative, aerobic, orange-pigmented bacterial strain, designated HHU K3-1 T, was isolated from the surface water of the Yellow Sea. The strain was observed to grow on 2216E agar medium, and growth occurred at pH 6.0–8.0 (optimum 7.0), 28–37 °C (optimum 28 °C), and in the presence of 0.5–5% (w/v) NaCl (optimum 1–3%). The major fatty acids (> 10%) were summed feature 3 (C16:1ω6c/C16:1ω7c), C17:1ω6c and summed feature 8 (C18:1ω6c/C18:1ω7c). Strain HHU K3-1 T was found to contain ubiquinone-10 as the predominant quinone and the major polar lipids were diphosphatidylglycerol (DPG), phosphatidylethanolamine (PE), phosphatidylglycerol (PG) and sphingoglycolipid (SGL). The 16S rRNA gene sequence analysis indicated that strain HHU K3-1 T shared highest similarities with Pelagerythrobacter marensis KCTC 22370 T (97.7%) and Qipengyuania oceanensis MCCC 1A09965T (96.9%). However, a phylogenetic tree based on 288 orthologous clusters (OCs) indicated that HHU K3-1 T was close related to Parapontixanthobacter aurantiacus MCCC 1A09962T. The pairwise AAI and evolutionary distance between HHU K3-1 T and Parapontixanthobacter aurantiacus MCCC 1A09962T are 67.1% and 0.43, respectively, which meet the recently proposed standard to differentiate genera in the family Erythrobacteraceae. On the basis of the result obtained by the polyphasic taxonomic study, strain HHU K3-1 T can be considered to represent a novel genus in the family Erythrobacteraceae, for which the name Actirhodobacter atriluteus gen. nov., sp. nov. is proposed. The type strain is HHU K3-1 T (= MCCC 1K04225T = KCTC 72834 T = CGMCC 1.17395 T).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The family Erythrobacteraceae belongs to the order Sphingomonadales of the class Alphaproteobacteria in the phylum Proteobacteria (Kosako et al. 2000). At the time of writing, the number of validly published genera of this family has reached 17 including the original 6 ones and the recently proposed 11 ones, Alteraurantiacibacter, Altererythrobacter, Altericroceibacterium, Alteripontixanthobacter (Apb.), Alteriqipengyuania Aurantiacibacter, Croceibacterium, Croceicoccus Erythrobacter, Parapontixanthobacter (Ppb.), Paraurantiacibacter, Parerythrobacter, Pelagerythrobacter (Pel), Pontixanthobacter, Pseudopontixanthobacter, Qipengyuania and Tsuneonella, according to the genus boundaries of average amino acid identity (AAI, 70%) and evolutionary distance (0.40) recently proposed for the family Erythrobacteraceae (Xu et al. 2020; Sun et al. 2020). Bacteria of the family Erythrobacteraceae were isolated from various habitats, including air (Xue et al. 2016), soil (Li et al. 2020), sediment (Park et al. 2019b), tidal flat (Park et al. 2019a), seawater (Li et al. 2017a; Meng et al. 2019), estuary (Park et al. 2019c) and freshwater (Kang et al. 2016). In addition, they also exist in some plant (Shiba and Simidu 1982), animal (Zhuang et al. 2019) species and some extreme environment, such as mudstone core (Zhang et al. 2016), hot spring (Yuan et al. 2017) and desert (Yan et al. 2017). Members of the family Erythrobacteraceae present pink, red, yellow or orange colonies on the media, and the cells are Gram-stain-negative, coccoid, ovoid or rod-shaped, and taking ubiquinone-10 as the dominating quinone (Tonon et al. 2014). Most members contain carotenoids (Huang et al. 2015), and several members could biosynthesize bacteriochlorophyll a (BChl a) (Shiba and Simidu 1982; Yurkov et al. 1994), which are also known as aerobic anoxygenic phototrophic bacteria (AAPB), can harvest light energy, and play a significant role in the carbon cycling of the oceans globally (Li et al. 2017b).
Hereby, we report a bacterium of the family Erythrobacteraceae, designated HHU K3-1 T, which was isolated from the surface water of the Yellow Sea of China. On the basis of data from the average nucleotide identity (ANI) values, digital DNA–DNA hybridization (dDDH), AAI and evolutionary distance between strain HHU K3-1 T and closely related members of the family Erythrobacteraceae, strain HHU K3-1 T should be considered to represent a novel genus within the family Erythrobacteraceae.
Material and methods
Bacteria isolate and culture conditions
Strain HHU K3-1 T was isolated from a surface water sample collected from the Yellow Sea of China (32°30.11′N, 122°18.89′E). Generally, 0.1 mL sea water was spread on 2216E medium (Solarbio, China) immediately after the water sample was collected. The plate was incubated at 28 °C for 7 days, and single colonies were selected and cultivated on 2216E plates. The strain HHU K3-1 T was maintained on 2216E medium and stored as aqueous glycerol suspensions (20%, v/v) at −80 °C. Ppb. aurantiacus MCCC 1A09962T and Pel. marensis KCTC 22370 T were used as reference strains for morphological, physiological, biochemical and chemotaxonomic analyses under the same experimental conditions as strain HHU K3-1 T. Strains Ppb. aurantiacus MCCC 1A09962T and Pel. marensis KCTC 22370 T were provided by the Marine Culture Collection of China (MCCC) and Korean Collection for Type Cultures (KCTC), respectively.
Phylogenetic and genome sequence analysis
Genomic DNA of HHU K3-1 T was extracted by using the Ezup Column Bacteria Genomic DNA Purification Kit (Sangon Biotech, China) according to the manufacturer’s instructions. The 16S rRNA gene was amplified with universal bacterial primers 27F and 1492R as described previously (Weisburg et al. 1991). The analysis of the 16S rRNA gene sequences of the strains was performed on the EzBioCloud server (https://www.ezbiocloud.net/) (Yoon et al. 2017). As for phylogenetic analysis, the neighbor-joining (NJ) (Saitou and Nei 1987), maximum-likelihood (ML) (Felsenstein 1981) and maximum-parsimony (MP) (Fitch 1971) methods were used to construct phylogenetic tree in the MEGA X software package (Kumar et al. 2018) after multiple alignments of sequences. Kimura’s two parameter model (Kimura 1980) was used to calculate evolutionary distance matrices of the phylogenetic trees. A bootstrap analysis with 1,000 replicates was also performed (Felsenstein 1985).
Genome sequencing of strain HHU K3-1 T was carried out at Magigene company, Guangzhou, China. Genome assembly and gap filling were done using SPAdes version 3.13.0 (Nurk et al. 2013) in UGENE software package (Okonechnikov et al. 2012). The ANI based on BLAST (OrthoANIb) was calculated on the EzBioCloud server (https://www.ezbiocloud.net/) (Richter and Rossello-Mora 2009; Yoon et al. 2017). The digital DNA–DNA hybridization (dDDH) values were calculated using the Genome-to-Genome Distance Calculator (GGDC) available at https://ggdc.dsmz.de (Meier-Kolthoff et al. 2013). Formula 2 was used for the dDDH analysis. AAI value was calculated on web server (http://enve-omics.ce.gatech.edu/aai/) (Qin et al. 2014). The whole genome sequences of the strains were annotated using the NCBI Prokaryotic Genome Annotation Pipeline (Tatusova et al. 2016) and eggNOG online server (https://eggnog-mapper.embl.de/) (Huerta-Cepas et al. 2017). DNA G + C content was determined according to the genome sequences.
In order to construct a robust phylogeny, a tree based on the concatenation of the 288 orthologous clusters (OCs) defined among the family Erythrobacteraceae in our previous study (Xu et al. 2020), was inferred by using EasyCGTree software package (https://github.com/zdf1987/EasyCGTree) under Windows operation system (OS), of which the algorithm was originally introduced by Zhang et al. (2020). EasyCGTree under Windows OS integrates blast + (Altschul et al. 1997) for gene calling, muscle (Edgar 2004) for sequence alignment and FastTree (Price et al. 2009) for phylogeny construction, while it employs Clustal Omega (Sievers et al. 2011) instead for sequence alignment under Linux OS. The protein sequences of the 288 OCs from Ppb. aurantiacus MCCC 1A09962T (Assembly No. GCA_009827635.1) were retrieve from our previous study (Xu et al. 2020) and used as reference for homolog searching of strain HHU K3-1 T. The evolutionary distance was calculated by using IQ-Tree 1.6.1 software (Nguyen et al. 2015) and LG + F + R9 substitution models, based on the concatenated protein sequences of the 288 OCs defined among the family Erythrobacteraceae in our previous study (Xu et al. 2020).
Morphology, physiology, and biochemical analysis
Gram staining was performed using the Gram Stain kit (G1060, Solarbio, China) according to the manufacturer’s instructions. The temperature range for growth were tested at 4, 10, 20, 28, 37 42 °C on 2216E plates. The salt tolerance range were tested at different NaCl concentrations (0%, 0.5%, 1%, 2%, 3%, 5%, 7%, 10% and 12%, w/v) and pH tolerance range were determined at different pH (pH 4.0–10.0, at intervals of 1.0 unit) in 2216E medium. Motility was determined by observing growth of cells in test tubes containing semisolid 2216E medium with 0.5% agar after 3 days of incubation at 28 °C (Cowan and Steel 1996). Other physiological and biochemical characterization were performed using the Biolog GEN III microtest system (Biolog, USA), API 20NE and API ZYM systems (bioMérieux, France) according to the manufacturer’s instructions.
Chemotaxonomic characterization
Cells of strain HHU K3-1 T and the reference type strains were harvested from cultures grown on 2216E liquid medium for 2 days at 28 °C. Cellular fatty acids were extracted and analyzed according to the method described by the Sherlock Microbial Identification (Sasser 1990). Respiratory quinone was purified (Hiraishi et al. 1996) and analyzed by HPLC (Tamaoka 1986). Polar lipids were examined by using two dimensional thin-layer chromatography according to the previously described method (Collins and Jones 1981; Minnikin et al. 1984).
Results and discussion
Genomic features and phylogenetic analysis
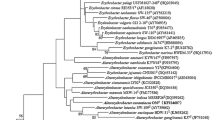
Based on the analysis of the 16S rRNA gene sequences on EzBioCloud server, strain HHU K3-1 T should belonged to the family Erythrobacteraceae and showed highest similarity with Pel. marensis KCTC 22370 T (97.6%), which was the only taxon with a similarity > 97%. Phylogenetic trees based on 16S rRNA gene using NJ, ML and MP algorithms, all indicated that strain HHU K3-1 T clustered with Ppb. aurantiacus MCCC 1A09962T (16S rRNA gene similarity 96.7%) and Apb. maritimus HME 9302 T (95.4%) (Fig. 1, S1 and S2). However, this phylogeny was not well supported (bootstrap value < 70%) in all three trees.

Neighbour-joining phylogenetic tree showing the relationship between strain HHU K3-1 T and its closest relatives. The black dots indicate branches that were also recovered using the maximum-parsimony and maximum-likelihood methods. Bootstrap values (expressed as percentages of 1000 replications) of above 70% are shown at the branch points nodes. The GenBank accession numbers are indicated in the brackets at the end of the tip labels. Sphingomonas paucimobilis ATCC 29837 T is used as out group. Bar, 0.02 substitutions per nucleotide position
Whole Genome Shotgun project of strain HHU K3-1 T has been performed and the genome sequence was deposited at GenBank/EMBL/DDBJ under the accession number JABWGV000000000. The draft genome sequence of strain HHU K3-1 T was 2,939,611 bp in length with 23 contigs, and the genome G + C content was 62.1%. And there was no photosynthesis gene cluster (PGC) present in the genome based on eggNOD online annotation result. As for the phylogenetic tree based on the 288 OCs, all the clades were well supported (bootstrap value > 70%) (Fig. 2). Different from the phylogenetic relationships based on 16S rRNA gene, strain HHU K3-1 T formed a clade with Ppb. aurantiacus MCCC 1A09962T, while Apb. maritimus HME 9302 T and Pel. marensis KCTC 22370 T were distant. Such a disagreement between phylogenetic trees based on 16S rRNA gene and core/house-keeping gene sets in the family Erythrobacteraceae have been well characterized previously (Xu et al. 2020), and the trees based on core/house-keeping gene sets are considered more reliable. In this study, the tree of the family Erythrobacteraceae based on the 288 OCs showed a very similar topology with the tree based on 288 single-copy orthologous clusters (Xu et al. 2020). The ANI value between strain HHU K3-1 T and Pel. marensis KCTC 22370 T was 73.6% (Table 1), which was significantly lower than the threshold value of the species boundary (95–96%) (Richter and Rossello-Mora 2009). While, dDDH value (results from the recommended formula 2) between strain HHU K3-1 T and Pel. marensis KCTC 22370 T was 18.5% (Table 1), which is also lower than the proposed and generally accepted species boundaries of 70% (Goris et al. 2007). For the family Erythrobacteraceae, it was proposed that the AAI value of < 70% and evolutionary distance of > 0.4 based on the 288 OCs be used to define different genera (Xu et al. 2020). Therefore, we further analyzed the pairwise AAI and evolutionary distance between strain HHU K3-1 T and close related taxa as previously descripted method (Xu et al. 2020). The AAI values between HHU K3-1 T and Ppb. aurantiacus MCCC 1A09962T, Apb. maritimus HME 9302 T and Pel. marensis KCTC 22370 T were 67.1%, 67.2% and 68.5%, respectively, while the evolutionary distances were 0.43, 0.68 and 0.61, respectively. According to the analysis of phylogeny, ANI, dDDH, AAI and evolutionary distance, strain HHU K3-1 T should represent a novel genus in the family Erythrobacteraceae.

Maximum-likelihood phylogenetic tree based on the 288 orthologous clusters (OCs) showing the position of strain HHU K3-1 T. Bootstrap values are shown at the branch points nodes. GenBank accession number or whole genome shotgun (WGS) project number is indicated in the bracket. Rhodospirillum rubrum ATCC 1170 T is used as out group. Bar, 0.1 substitutions per nucleotide position
Physiology, and biochemical analysis
Cells of strain HHU K3-1 T were Gram-staining negative, aerobic, nonmotile, catalase- and oxidase-positive. Optimal growth was observed at pH 7.0, at 28 °C and with 2% (w/v) NaCl. In the API ZYM tests, HHU K3-1 T was positive for acid phosphatase, alkaline phosphatase, esterase (C4), esterase lipase (C8), leucine arylamidase, naphthol-AS-BI-phosphohydrolase and α-chymotrypsin, but negative for cystine arylamidase, lipase (C14), N-acetyl-β-glucosaminidase, trypsin, valine arylamidase, α-fucosidase, α-galactosidase, α-glucosidase, α-mannosidase, β-galactosidase, β-glucosidase and β-glucuronidase. In the API 20NE tests, HHU K3-1 T was only positive for hydrolysis of esculin, but negative for arginine dihydrolase, D-glucose fermentation, indole production, hydrolysis of urea and gelatin, nitrate reduction and 4-nitrophenyl β-D-galactopyranoside. As for assimilation, HHU K3-1 T could not assimilate adipic acid, capric acid, D-glucose, D-maltose, D-mannitol, D-mannose, L-arabinose, malic acid, N-acetylglucosamine, phenylacetic acid, potassium gluconate, and trisodium citrate. In the BIOLOG GEN III tests, HHU K3-1 T was positive for utilization of acetic acid, acetoacetic acid, L-glutamic acid, tween 40, was resistant to nalidixic acid, and could utilize L-pyroglutamic acid (Table 2).
Chemotaxonomic characterization
The total cellular fatty acids of strain HHU K3-1 T and its most related species were shown in Table S1. The predominant fatty acids (> 10% of the total fatty acids) of strain HHU K3-1 T were summed feature 8 (C18:1 ω7c/C18:1 ω6c) (33.5%), C17:1 ω6c (24.0%) and summed feature 3 (C16:1 ω7c/C16:1 ω6c) (12.3%). The major polar lipid of strain HHU K3-1 T were diphosphatidylglycerol (DPG), phosphatidylethanolamine (PE), phosphatidylglycerol (PG) and sphingoglycolipid (SGL), which is similar to those of the closely related type strains Ppb. aurantiacus MCCC 1A09962T and Pel. marensis KCTC 22370 T. In addition, two unknown phospholipid and one unidentified lipid were also detected. The major respiratory quinone of strain HHU K3-1 T was ubiquinone-10, which is common in the species of the genus Altererythrobacter and other genera (Feng et al. 2015).
Conclusion
The ANIb and dDDH analysis between HHU K3-1 T and close related species were all below the generally accepted species boundaries 95%-96% and 70%, respectively (Table 1). The AAI analysis between HHU K3-1 T and close related genera also showed pairwise AAI values of < 70% and evolutionary distances of > 0.40, which was proposed to define different genera in the family Erythrobacteraceae (Xu et al. 2020). The major fatty acids (> 10%) of strain HHU K3-1 T were summed feature 8 (C18:1ω7c/C18:1ω6c), summed feature 3 (C16:1ω7c/C16:1ω6c) and C17:1ω6c (Table S1). Nevertheless, those of Ppb. aurantiacus MCCC 1A09962T were summed feature 3 (C16:1ω7c/C16:1ω6c), C16:0 and summed feature 8 (C18:1ω7c/C18:1ω6c), and those of Pel. marensis KCTC 22370 T were summed feature 8 (C18:1ω7c/C18:1ω6c) and 11-methyl-C18:1ω7c. Strain HHU K3-1 T contained respiratory ubiquinone-10 as the main respiratory quinone, which was consistent with the other members of the family Erythrobacteraceae. As for polar lipids, strain HHU K3-1 T and the close related genera all contained diphosphatidylglycerol (DPG), phosphatidylethanolamine (PE), phosphatidylglycerol (PG) and sphingoglycolipid (SGL). On the basis of phylogenetic, phenotypic and chemotaxonomic data, strain HHU K3-1 T should be proposed as a novel genus within the family Erythrobacteraceae, for which the name Actirhodobacter atriluteus gen. nov., sp. nov. is proposed.
Description of Actirhodobacter gen. nov.
Actirhodobacter (Ac.ti.rho.do.bac’ter. L. fem. n. acta, seaside, shore; Gr. neut. n. rhodon, the rose; N.L. masc. n. bacter, rod; N.L. masc. n. Actirhodobacter, red-colored rod from the seaside).
Cells are Gram-staining-negative, non-motile, aerobic and have no soluble pigments. Colonies are orange, smooth and opaque. The dominant respiratory quinone is ubiquinone-10. The major polar lipids are diphosphatidylglycerol (DPG), phosphatidylethanolamine (PE), phosphatidylglycerol (PG) and sphingoglycolipid (SGL). The genomic G + C content is 62.1%. As determined by 16S rRNA gene sequence analysis, the genus is a member of the family Erythrobacteraceae, class Alphaproteobacteria. The type species is Actirhodobacter atriluteus.
Description of Actirhodobacter atriluteus sp. nov.
Actirhodobacter atriluteus (at.ri.lu’te.us. L. mas. adj. ater dark; L. masc. adj. luteus yellow-orange; N.L. masc. adj. atriluteus dark orange).
Cells are Gram-stain-negative, non-motile and aerobic. After incubation on 2216E agar for 3 days. Colonies are orange, smooth and opaque. Growth occurs at 28–37 °C (optimum 28 °C), at pH 6.0–8.0 (optimum pH 7.0) and in the presence of NaCl concentrations of 0.5–5% (optimum 1–3%). The predominant respiratory quinone is ubiquinone-10, The major polar lipids are diphosphatidylglycerol (DPG), phosphatidylethanolamine (PE), phosphatidylglycerol (PG) and sphingoglycolipid (SGL), two unknown phospholipid and one unidentified lipid. The predominant fatty acids (> 10%) were summed feature 8 (C18:1ω7c/C18:1ω6c), C17:1ω6c and summed feature 3 (C16:1ω7c/C16:1ω6c).
The type strain, HHU K3-1 T (= MCCC 1K04225T = KCTC 72834 T = CGMCC 1.17395 T), was isolated from the surface water of Yellow Sea, China. The DNA G + C content is 62.1%.
The 16S rRNA gene sequence has been deposited at GenBank/EMBL/DDBJ accession number MW172823, while the genome sequence has been deposited at GenBank/EMBL/DDBJ under accession number JABWGV000000000.
Availability of data and materials
Most of the data supporting the conclusions of this article are included within the article and its additional files. The genome datasets and the 16S rRNA gene sequence of Actirhodobacter atriluteus HHU K3-1 T generated during the current study are available in the GenBank repository under accession number JABWGV000000000 and MW172823, respectively. Other datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Altschul SF, Madden TL, Schäffer AA et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. https://doi.org/10.1093/nar/25.17.3389
Collins MD, Jones D (1981) A note on the separation of natural mixtures of bacterial ubiquinones using reverse-phase partition thin-layer chromatography and high performance liquid chromatography. J Appl Bacteriol 51:129–134. https://doi.org/10.1111/j.1365-2672.1981.tb00916.x
Cowan ST, Steel KJ (1996) Manual for the identification of medical bacteria. Cambridge University Press, London, p 232
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform 5:1–19. https://doi.org/10.1186/1471-2105-5-113
Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17:368–376. https://doi.org/10.1007/bf01734359
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791. https://doi.org/10.1111/j.1558-5646.1985.tb00420.x
Feng X-M, Mo Y-X, Han L, Nogi Y, Zhu Y-H, Lv J (2015) Qipengyuania sediminis gen. nov., sp. nov., a member of the family Erythrobacteraceae isolated from subterrestrial sediment. Int J Syst Evol Microbiol 65:3658–3665. https://doi.org/10.1099/ijsem.0.000472
Fitch WM (1971) Toward defining the course of evolution: minimum change for a specific tree topology. Syst Biol 20:406–416. https://doi.org/10.2307/2412116
Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM (2007) DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57:81–91. https://doi.org/10.1099/ijs.0.64483-0
Hiraishi A, Ueda Y, Ishihara J, Mori T (1996) Comparative lipoquinone analysis of influent sewage and activated sludge by high-performance liquid chromatography and photodiode array detection. J Gen Appl Microbiol 42:457–469. https://doi.org/10.2323/jgam.42.457
Huang Y, Zeng Y, Feng H, Wu Y, Xu X (2015) Croceicoccus naphthovorans sp. nov., a polycyclic aromatic hydrocarbons-degrading and acylhomoserine-lactone-producing bacterium isolated from marine biofilm, and emended description of the genus Croceicoccus. Int J Syst Evol Microbiol 65:1531–1536. https://doi.org/10.1099/ijs.0.000132
Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, von Mering C, Bork P (2017) Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol Biol Evol 34:2115–2122. https://doi.org/10.1093/molbev/msx148
Kang JW, Kim MS, Lee JH, Baik KS, Seong CN (2016) Altererythrobacter rigui sp. nov., isolated from wetland freshwater. Int J Syst Evol Microbiol 66:2491–2496. https://doi.org/10.1099/ijsem.0.001078
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120. https://doi.org/10.1007/bf01731581
Kosako Y, Yabuuchi E, Naka T, Fujiwara N, Kobayashi K (2000) Proposal of Sphingomonadaceae fam, nov., consisting of Sphingomonas Yabuuchi et al, 1990, Erythrobacter Shiba and Shimidu 1982, Erythromicrobium Yurkov et al, 1994, Porphyrobacter Fuerst et al, 1993, Zymomonas Kluyver and van Niel 1936, and Sandaracinobacter Yurkov et al, 1997, with the type genus Sphingomonas Yabuuchi et al, 1990. Microbiol Immunol 44:563–575. https://doi.org/10.1111/j.1348-0421.2000.tb02535.x
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Li D-D et al (2017a) Erythrobacter xanthus sp. nov., isolated from surface seawater of the South China Sea. Int J Syst Evol Microbiol 67:2459–2464. https://doi.org/10.1099/ijsem.0.001991
Li Q, Song A, Peng W, Jin Z, Mueller WEG, Wang X (2017b) Contribution of aerobic anoxygenic phototrophic bacteria to total organic carbon pool in aquatic system of subtropical karst catchments, Southwest China: evidence from hydrochemical and microbiological study. Fems Microbiol Ecol 93:6. https://doi.org/10.1093/femsec/fix065
Li H-P, Yao D, Shao K-Z, Han Q-Q, Gou J-Y, Zhao Q, Zhang J-L (2020) Altererythrobacter rhizovicinus sp. nov., isolated from rhizosphere soil of Haloxylon ammodendron. Int J Syst Evol Microbiol 70:680–686. https://doi.org/10.1099/ijsem.0.003817
Meier-Kolthoff JP, Auch AF, Klenk H-P, Goeker M (2013) Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform 14:60. https://doi.org/10.1186/1471-2105-14-60
Meng F-X et al (2019) Altererythrobacter aerophilus sp. nov., isolated from deep-sea water of the north-west Pacific. Int J Syst Evol Microbiol 69:1689–1695. https://doi.org/10.1099/ijsem.0.003377
Minnikin DE et al (1984) An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J Microbiol Meth 2:233–241. https://doi.org/10.1016/0167-7012(84)90018-6
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274. https://doi.org/10.1093/molbev/msu300
Nurk S et al (2013) Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol 20:714–737. https://doi.org/10.1089/cmb.2013.0084
Okonechnikov K, Golosova O, Fursov M, Team U (2012) Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28:1166–1167. https://doi.org/10.1093/bioinformatics/bts091
Park S, Park J-M, Oh T-K, Yoon J-H (2019a) Altererythrobacter insulae sp. nov., a lipolytic bacterium isolated from a tidal flat. Int J Syst Evol Microbiol 69:1009–1015. https://doi.org/10.1099/ijsem.0.003260
Park S, Park J-M, Yoon J-H (2019b) Altererythrobacter aquimixticola sp. nov., isolated from sediment sampled at the junction between the ocean and a freshwater spring. Int J Syst Evol Microbiol 69:2408–2414. https://doi.org/10.1099/ijsem.0.003494
Park S, Won SM, Yoon J-H (2019c) Erythrobacter marisflavi sp. nov., isolated from isolated from estuary water. Int J Syst Evol Microbiol 69:2696–2702. https://doi.org/10.1099/ijsem.0.003510
Price MN, Dehal PS, Arkin AP (2009) FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26:1641–1650. https://doi.org/10.1093/molbev/msp077
Qin Q-L et al (2014) A proposed genus boundary for the prokaryotes based on genomic insights. J Bacteriol 196:2210–2215. https://doi.org/10.1128/jb.01688-14
Richter M, Rossello-Mora R (2009) Shifting the genomic gold standard for the prokaryotic species definition. P Natl Acad Sci USA 106:19126–19131. https://doi.org/10.1073/pnas.0906412106
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425. https://doi.org/10.1093/oxfordjournals.molbev.a040454
Sasser M (1990) Identification of bacteria by gas chromatography of cellular fatty acids, MIDI Technical Note 101. Microbial ID, Inc, Newark
Shiba T, Simidu U (1982) Erythrobacter longus gen. nov., sp. nov., an aerobic bacterium which contains bacteriochlorophyll a. Int J Syst Bacteriol 32:211–217. https://doi.org/10.1099/00207713-32-2-211
Sievers F et al (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol Syst Biol 7:539. https://doi.org/10.1038/msb.2011.75
Sun J, Guo J, Lin T-H, Feng X, Zhang R (2020) Pseudopontixanthobacter vadosimaris gen. nov., sp. nov., isolated from shallow sea near Kueishan Island. Int J Syst Evol Microbiol 70:6444–6449. https://doi.org/10.1099/ijsem.0.004552
Tamaoka J (1986) Analysis of bacterial menaquinone mixtures by reverse-phase high-performance liquid chromatography. Meth Enzymol 123:251–256. https://doi.org/10.1016/s0076-6879(86)23028-1
Tatusova T et al (2016) NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 44:6614–6624. https://doi.org/10.1093/nar/gkw569
Tonon LAC, Moreira APB, Thompson F (2014) The Family Erythrobacteraceae. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (eds) The prokaryotes: Alphaproteobacteria and Betaproteobacteria. Springer, Berlin, pp 213–235. https://doi.org/10.1007/978-3-642-30197-1
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703. https://doi.org/10.1128/jb.173.2.697-703.1991
Xu L, Sun C, Fang C, Oren A, Xu X-W (2020) Genomic-based taxonomic classification of the family Erythrobacteraceae. Int J Syst Evol Microbiol 70:4470–4495. https://doi.org/10.1099/ijsem.0.004293
Xue H, Piao C-g, Guo M-w, Wang L-f, Fang W, Li Y (2016) Description of Altererythrobacter aerius sp. nov., isolated from air, and emended description of the genus Altererythrobacter. Int J Syst Evol Microbiol 66:4543–4548. https://doi.org/10.1099/ijsem.0.001388
Yan Z-F, Lin P, Won K-H, Yang J-E, Li C-T, Kook M, Yi T-H (2017) Altererythrobacter deserti sp. nov., isolated from desert soil. Int J Syst Evol Microbiol 67:3806–3811. https://doi.org/10.1099/ijsem.0.002197
Yoon S-H, Ha S-M, Kwon S, Lim J, Kim Y, Seo H, Chun J (2017) Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol 67:1613–1617. https://doi.org/10.1099/ijsem.0.001755
Yuan C-G et al (2017) Altererythrobacter lauratis sp. nov. and Altererythrobacter palmitatis sp. nov., isolated from a Tibetan hot spring. Antonie Van Leeuwenhoek 110:1077–1086. https://doi.org/10.1007/s10482-017-0882-y
Yurkov V et al (1994) Phylogenetic positions of novel aerobic, bacteriochlorophyll a-containing bacteria and description of Roseococcus thiosulfatophilus gen. nov., sp. nov., Erythromicrobium ramosum gen. nov., sp. nov., and Erythrobacter litoralis sp. nov. Int J Syst Bacteriol 44:427–434. https://doi.org/10.1099/00207713-44-3-427
Zhang W, Yuan X, Feng Q, Zhang R, Zhao X, Lv J (2016) Altererythrobacter buctense sp. nov., isolated from mudstone core. Antonie Van Leeuwenhoek 109:793–799. https://doi.org/10.1007/s10482-016-0679-4
Zhang D-F, Cui X-W, Zhao Z, Zhang A-H, Huang J-K, Li W-J (2020) Sphingomonas hominis sp. nov., isolated from hair of a 21-year-old girl. Antonie Van Leeuwenhoek 113:1523–1530. https://doi.org/10.1007/s10482-020-01460-z
Zhuang L, Lin B, Xu L, Li G, Wu C-J, Luo L (2019) Erythrobacter spongiae sp. nov., isolated from marine sponge. Int J Syst Evol Microbiol 69:1111–1116. https://doi.org/10.1099/ijsem.0.003278
Funding
This research was supported by the National Natural Science Foundation of China (No. 31900001 and 32000001), the China Postdoctoral Science Foundation (2020M671312) and the Philosophy and Social Science Research Fund Project for Jiangsu Universities (2018SJA0116). Expenditures of sampling are funded by Project No. 31960001; expenditures of sequencing and polyphasic taxonomic experiences are funded by Project No. 32000001; expenditures of student service fees are funded by Project No. 2020M671312 and 2018SJA0116.
Author information
Authors and Affiliations
Contributions
DFZ designed research and project outline. HPX, DFZ, and LX performed isolation, deposition and polyphasic taxonomy. DFZ, XNW and HPX performed genome analysis. HPX, DFZ, XNW and AHZ drafted the manuscript. JKH and CL revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xue, HP., Zhang, DF., Xu, L. et al. Actirhodobacter atriluteus gen. nov., sp. nov., isolated from the surface water of the Yellow Sea. Antonie van Leeuwenhoek 114, 1059–1068 (2021). https://doi.org/10.1007/s10482-021-01576-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-021-01576-w