
Abstract
A Gram-stain-negative, aerobic, motile strain, HHU CXWT, was isolated from hair of a healthy 21-year-old female student of Hohai University, Nanjing, China. The 16S rRNA gene sequence analysis indicated that HHU CXWT represents a member of the genus Sphingomonas with the highest sequence similarity (97.6%) to the type strain S. aquatilis JSS7T. HHU CXWT grew at 4–35 °C and pH 6–8, with optimum growth at 28 °C and pH 7. Tolerance to NaCl was up to 2% (w/v) with optimum growth in 0.5–1.0% NaCl. The major fatty acids were C16:0, C17:1ω6c, C18:1ω7c11-methyl, summed feature 3 (C16:1ω7c and/or C16:1ω6c), and summed feature 8 (C18:1ω7c and/or C18:1ω6c). The predominant isoprenoid quinone was ubiquinone-10. The polar lipids were diphosphatidylglycerol (DPG), phosphatidylethanolamine (PE), phosphatidylglycerol (PG), sphingoglycolipid (SGL), phosphatidylinositol mannosides (PIM), and an unidentified glycolipid (GL). The DNA G + C content was 67.1%. The average nucleotide identity (ANI) values and digital DNA–DNA hybridization (dDDH) between HHU CXWT and closely related members of the genus Sphingomonas were all below the cut-off level (95–96% and 70%, respectively) for species delineation. On the basis of the phenotypic, phylogenetic and chemotaxonomic characterizations, HHU CXWT represents a novel species of the genus Sphingomonas, for which the name Sphingomonas hominis sp. nov. is proposed. The type strain is HHU CXWT (= KCTC 72946T = CGMCC 1.17504T = MCCC 1K04223T).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Members of Sphingomonas within Alphaproteobacteria have been isolated from a variety of environments, including human-associated niches, water (fresh and sea), endophytes, terrestrial habitats, sediment (river and subsurface), and rhizosphere soil (Asaf et al. 2020; Aylward et al. 2013). Many isolates possess multifaceted functions ranging from remediation of environmental contaminations to synthetizing highly beneficial phytohormones, such as polycyclic aromatic hydrocarbon (PAH) degradation and sphingan producing. Some species of the genus have also been noted to improve plant-growth during stress conditions such as drought, salinity, and heavy metals in agricultural soil, because of their potential to produce plant growth hormones e.g. gibberellins and indole acetic acid (Asaf et al. 2020; Khan et al. 2014; Yang et al. 2014)
Currently (June, 2020), the genus Sphingomonas includes 131 species with validly published and correct names according to the List of Prokaryotic names with Standing in Nomenclature (LPSN) (Parte 2018). A punctiform, orange strain designated HHU CXWT was isolated from the hair of a healthy 21-year-oldfemale student in class of microbiology experiment, Hohai University, Nanjing, China. The 16S rRNA gene analysis indicated strain HHU CXWT possibly represent a novel species in genus Sphingomonas, and polyphasic taxonomy was performed on this strain subsequently.
Materials and methods
Isolation of the bacterial strain and culture conditions
The hair was collected from a female student in class, and was cut into pieces (< 1 cm in length) before being bespattered on the trypticase soy agar (TSA) medium (BD Diagnostics, Maryland, USA). The plate was incubated at 28 °C for 5 days, and single colonies were selected and cultivated on TSA plates. The strain HHU CXWT was maintained on TSA medium and stored as aqueous glycerol suspensions (20%, v/v) at − 80 °C.
Phenotypic and biochemical characterization
Colony properties of strain HHU CXWT were observed on TSA medium. Cell morphology was examined using optical microscopy (Axio Vert A1, Zeiss, German) after 2-day-incubation in trypticase soy broth (TSB) (BD Diagnostics, Maryland, USA) at 28 °C with vibration (180 rpm). Gram staining was determined by using a Gram Stain kit (G1060, Solarbio, China) according to the manufacturer’s instructions. Growth was tested at 4, 10, 20, 28, 35 and 37 °C on TSA medium. The pH range for growth was determined by measuring the optical densities (at 600 nm) of TSB cultures after 2 days. The pH was adjusted to pH 4–10 (at intervals of 1.0 pH unit) in a vertical flow clean bench after sterilization, using NaOH and HCl solutions. NaCl tolerance was determined on TSA plates adjusted to 0%, 0.5%, 1%, 2%, 3%, 4% and 5% concentrations (w/v). Motility was determined by observing growth of cells in test tubes containing semisolid TSA medium with 0.5% agar after 3 days of incubation at 28 °C (Cowan and Steel 1996). Additional physiological and biochemical characterization were performed using the Biolog GEN III microtest system (Biolog, USA), API 20NE and API ZYM systems (bioMérieux, France) according to the manufacturer’s instructions.
Chemotaxonomic characterisation
The biomass used for analysis of cellular fatty acids, polar lipids, and quinones, were obtained from cultures grown in TSB medium for 2 days at 28 °C. Cellular fatty acids were extracted, methylated and analyzed by using the Sherlock Microbial Identification System (MIDI) according to previous method (Sasser 1990) and the manufacturer’s instructions. Quinones were extracted (Collins et al. 1977) and detected by HPLC (Tamaoka 1986). Polar lipids were determined according to published procedures (Collins and Jones 1980; Minnikin et al. 1979).
Phylogenetic and genotypic analysis
Genomic DNA was isolated by using the Ezup Column Bacteria Genomic DNA Purification Kit (Sangon Biotech, China), and PCR amplification of the 16S rRNA gene sequence were performed with the universal primers 27F and 1492R (Lane 1991). The obtained 16S rRNA gene sequence was analyzed on the EzBioCloud server (https://www.ezbiocloud.net/) (Yoon et al. 2017). Phylogenetic analysis was carried out based on the neighbor-joining (Saitou and Nei 1987), maximum-likelihood (Felsenstein 1981) and maximum-parsimony (Fitch 1971) methods by using the MEGA X software package (Kumar et al. 2018) after multiple alignments of sequences using Clustal version 2.1 program (Larkin et al. 2007). Evolutionary distance matrices of phylogenetic trees were calculated according to Kimura’s two-parameter model (Kimura 1980). Bootstrap analysis was performed with 1000 replications (Felsenstein 1985).
Whole-genome sequencing of HHU CXWT was performed using paired-end sequencing method with Hiseq X platform (Illumina) at Magigene Company, Guangzhou, PR China. Reads of each data set were filtered, and high-quality paired-end reads were assembled using SPAdes version 3.13.0 (Nurk et al. 2013) in UGENE software package (Okonechnikov et al. 2012). Sequences with coverage < 5 or length < 500 bp were excluded from the output genome sequences. The average nucleotide identity (ANI) based on BLAST (ANIb) was determined using JSpecies version 1.2.1 (Richter and Rossello-Mora 2009) while the digital DNA-DNA hybridization (dDDH) values were calculated using the Genome-to-Genome Distance Calculator (GGDC) available at https://ggdc.dsmz.de (Meier-Kolthoff et al. 2013). Formula 2 was applied for the dDDH analysis. Genome annotation was performed by the NCBI Prokaryotic Genome Annotation Pipeline (Tatusova et al. 2016) and eggNOD online server (https://eggnog-mapper.embl.de/) (Huerta-Cepas et al. 2017). The DNA G + C content of the genome was calculate based on the genome sequences.
A gene set called bac120 was used to infer a super phylogeny based on multilocus sequence analysis, which includes 120 universal single-copy genes among domain Bacteria (Parks et al. 2018). This genome-based tree was referred as ‘bac120 tree’. Generally, the protein sequences of bac120 from a S. ginsenosidimutans strain (Assembly No. GCF_002374835.1) were retrieve from the Genome Taxonomy Database (https://gtdb.ecogenomic.org/); local tblastn program in blast-2.9.0 + software package (Altschul et al. 1997) was used to search homologs in the Sphingomonas genomes of interest with 1e-5 evalue and 50% identity as cutoffs; the gene sequences of each gene cluster were aligned by using Clustal Omega version 1.2.1 (Sievers et al. 2011); a maximum-likelihood tree was inferred by applying FastTree 2.1 (Price et al. 2009) to the common genes of the bac120 set among all the genomes of interest after alignment trimming and concatenation. The bac120 tree was constructed on a set of genomes as input data, which was implement by using an in-house Perl script under Ubuntu operation system.
Results and discussion
Phylogenetic and genotypic characteristics
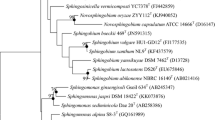
Based on the results from EzBioCloud server, HHU CXWT shared the highest 16S rRNA gene sequence similarity (99.1%) with two non-type strains Sphingomonas sp. JS21-1 (FNZZ01000011) and Sphingomonas sp. Mn802worker (AORY01000018) (Aylward et al. 2013), while S. aquatilis JSS7T (AF131295) from the valid published species of Sphingomona showed the highest similarity (97.6%) with strain HHU CXWT. All three phylogenetic trees using neighbor-joining, maximum-likelihood and maximum-parsimony algorithms, indicated that HHU CXWT clustered with Sphingomonas sp. JS21-1 and Sphingomonas sp. Mn802worker, forming a distinct but not well supported clade in the genus Sphingomona (Fig. 1, S1 and S2). Based on the 16S rRNA gene analysis, S. aquatilis KCTC 2881T (= JSS7T) was used as a reference for the subsequent tests.

Neighbour-joining phylogenetic tree based on 16S rRNA gene sequences showing the position of strain HHU CXWT. Bootstrap values (expressed as percentages of 1000 replications) of above 70% are shown at the branch nodes. The GenBank accession numbers are indicated in the brackets at the end of the tip labels. The black dots denote nodes that were also recovered using the maximum-likelihood and maximum-parsimony methods. Erythrobacter longus DSM 6997T is used as out group. Bar, 0.02 substitutions per nucleotide position
Whole Genome Shotgun project of HHU CXWT has been deposited at GenBank/EMBL/DDBJ under the accession number JABULH000000000. The draft genome consists of 36 contigs with a total size of 3,558,003 bp and genomic DNA G + C content 67.1%. A total of 3345 coding genes was predicted, of which 736, 1904 and 2802 genes were assigned to Gene Ontology (GO) (The Gene Ontology Consortium 2019), Kyoto Encyclopedia of Genes and Genomes (KEGG) (https://www.kegg.jp/) and Cluster of Orthologous Groups (COG) (Galperin et al. 2015) database, respectively. Future analysis of the KEGG annotation results from eggNOD server suggested that there were 28 genes associated with flagellar assembly (ko02040), 35 with biofilm formation (ko05111, ko02025 and ko02026), 7 with carotenoid biosynthesis (ko00361), respectively. Dramatically, strain HHU CXWT harbors a crtX gene (locus tag HRV97_09920), which could encode an enzyme catalyzing zeaxanthin to produce zeaxanthin diglucoside, and is rare present among Sphingomonadales (Fig. 2) (Siddaramappa et al. 2018).

Maximum-likelihood phylogenetic tree based on 69 common bac120 gene sequences showing the position of strain HHU CXWT. Bootstrap values of 100% are marked with black dots at the branch nodes. Tip labels followed by a asterisk indicated a crtX homolog was found in the genome, and the GenBank accession number or whole genome shotgun (WGS) project number is indicated in the bracket. Erythrobacter longus DSM 6997 T is used as out group. Bar, 0.05 substitutions per nucleotide position
According to the taxa of 16S rRNA gene tree, 15 Sphingomonas genomes together with genomes of strain HHU CXWT and Erythobacter longus DSM 6997T (JMIW01), were used to infer a robust tree based on the bac120 gene set, which was named bac120 tree in this study. A total of 69 genes from bac120 gene set was detected in all the 17 genomes and used in further analysis. The bac120 tree was well supported on all the branches (Fig. 2). The same as shown in the 16S rRNA gene tree, strains HHU CXWT is close related to isolates Sphingomonas sp. JS21-1 and Sphingomonas sp. Mn802worker. Additionally, the bac120 tree clarified that S. phyllosphaerae (Huang et al. 2012; Rivas et al. 2004) and S. adhaesiva (Feng et al. 2018) were the currently known valid species sharing the most recent ancestor with strain HHU CXWT.
The ANIb values between HHU CXWT and the close related strains Sphingomonas sp. JS21-1 (FNZZ01), Sphingomonas sp. Mn802worker (AORY01) and S. aquatilis NBRC 16722T (BJXI01) were all below 85%, while the dDDH values were 28.5%, 27.9%, and 21.1%, respectively (Table 1). These values are lower than the proposed and generally accepted species boundaries of 70% for dDDH and 95–96% for ANI (Goris et al. 2007; Richter and Rossello-Mora 2009), which suggest that strain HHU CXWT represents a novel species of the genus Sphingomona. Furthermore, Sphingomonas sp. JS21-1 and Sphingomonas sp. Mn802workers should represent two novel species of Sphingomonas, too, according to the ANIb and dDDH analysis (Table 1).
Phenotypic characteristics
Cells of HHU CXWT were Gram-staining negative, motile, aerobic and rod shaped. The colonies on TSA medium were orange, circular, smooth, and opaque. Growth of HHU CXWT was observed at 4–35 °C (optimum temperature, 28 °C) and at pH 6–8 (optimum, pH 7). The tolerance to NaCl was up to 2% (w/v) with optimum growth in the presence of 0.5–1.0% NaCl. HHU CXWT was catalase-positive and oxidase-negative. In the API ZYM tests, HHU CXWT was positive for acid phosphatase, alkaline phosphatase, cystine arylamidase, esterase (C4), esterase lipase (C8), leucine arylamidase, naphthol-AS-BI-phosphohydrolase, trypsin, valine arylamidase, and α-glucosidase, but negative for lipase (C14), N-acetyl-β-glucosaminidase, α-chymotrypsin, α-fucosidase, α-galactosidase, α-mannosidase, β-galactosidase, β-glucuronidase, and β-glucosidase. In the API 20NE tests, HHU CXWT was positive for hydrolysis of esculin and nitrate reduction, but negative for arginine dihydrolase, d-glucose fermentation, indole production, and hydrolysis of 4-nitrophenyl β-D-galactopyranoside, gelatin, and urea. Additionally, HHU CXWT was negative for assimilation of adipic acid, capric acid, d-glucose, d-maltose, d-mannitol, d-mannose, l-arabinose, malic acid, N-acetylglucosamine, phenylacetic acid, potassium gluconate, and trisodium citrate. In the BIOLOG GEN III tests, HHU CXWT was negative for all the tests, while the reference strain S. aquatilis KCTC 2881T was positive for utilization of α-d-glucose, glycyl-l-prolin, d-glucuronic acid, and l-glutamic acid, and was resistant to pH 6, 4% NaCl, lincomycin, and tetrazolium blue.
Chemotaxonomic characteristics
The major fatty acids (> 5%) of strain HHU CXWT were C16:0, C17:1ω6c, C18:1ω7c11-methyl, summed feature 3 (C16:1ω7c and/or C16:1ω6c), and summed feature 8 (C18:1ω7c and/or C18:1ω6c) (Table S1). The predominant isoprenoid quinone detected in strain HHU CXWT was ubiquinone-10, which is the typical quinone reported for the genus Sphingomonas (Li et al. 2019). The polar lipids of strain HHU CXWT mainly consisted of diphosphatidylglycerol (DPG), phosphatidylethanolamine (PE), phosphatidylglycerol (PG), sphingoglycolipid (SGL), and phosphatidylinositol mannosides (PIM), which is similar to that of S. aquatilis KCTC 2881T (Fig. S3). Minor amount of phosphatidylcholine (PC) and an unidentified polar lipid (L) were also detected.
Taxonomic conclusions
Strain HHU CXWT shared < 98% similarities of 16S rRNA gene with valid described species and formed a distinct clade in the genus Sphingomonas (Fig. 1, S1, and S2), and the ANIb and dDDH analysis between HHU CXWT and closely related species were all below the generally accepted species boundaries 95–96% and 70%, respectively (Table 1). Furthermore, strain HHU CXWT showed differences from closely related type strains S. aquatilis KCTC 2881 T and S. phyllosphaerae FA2T on colony color, nitrate reduction, assimilation of several carbon resource, diagnostic phospholipids and major fatty acids (Table 2). Based on the genotypic, phenotypic and chemotaxonomic features, strain HHU CXWT can be considered as a novel species of the genus Sphingomonas, for which the name Sphingomonas hominis sp. nov. is proposed.
Description of Sphingomonas hominis sp. nov.
Sphingomonas hominis (ho’mi.nis L. gen. masc. n. hominis, of a human being, named for the host from whose hair this species is found).
Cells are Gram-staining negative, aerobic, rod shaped, and motile. Colonies are orange, circular, smooth and opaque on TSA medium. Growth occurs at 4–35 °C (optimum temperature, 28 °C) and at pH 6–8 (optimum, pH 7). The tolerance to NaCl was up to 2% (w/v) with optimum concentration 0.5–1.0%. Catalase-positive and oxidase-negative. In the API ZYM tests, positive for acid phosphatase, alkaline phosphatase, cystine arylamidase, esterase (C4), esterase lipase (C8), leucine arylamidase, naphthol-AS-BI-phosphohydrolase, trypsin, valine arylamidase, and α-glucosidase; but negative for lipase (C14), N-acetyl-β-glucosaminidase, α-chymotrypsin, α-fucosidase, α-galactosidase, α-mannosidase, β-galactosidase, β-glucosidase, and β-glucuronidase. In the API 20NE tests: positive for nitrate reduction and hydrolysis of esculin; negative for arginine dihydrolase, d-glucose fermentation, indole production, and hydrolysis of 4-nitrophenyl β-D-galactopyranosidegelatin, and urea; negative for assimilation of adipic acid, capric acid, d-glucose, d-maltose, d-mannitol, d-mannose, l-arabinose, malic acid, N-acetylglucosamine, phenylacetic acid, potassium gluconate, and trisodium citrate. The major fatty acids are C16:0, C17:1ω6c, C18:1ω7c11-methyl, summed feature 3 (C16:1ω7c and/or C16:1ω6c), and summed feature 8 (C18:1ω7c and/or C18:1ω6c). The predominant isoprenoid quinone is ubiquinone-10. The polar lipids are diphosphatidylglycerol (DPG), phosphatidylethanolamine (PE), phosphatidylglycerol (PG), sphingoglycolipid (SGL), phosphatidylinositol mannosides (PIM), and an unidentified glycolipid (GL).
The type strain, HHU CXWT (= KCTC 72946T = CGMCC 1.17504T = MCCC 1K04223T), was isolated from the hair of a 21-year old female student of Hohai University, Nanjing, China. The GenBank/EMBL/DDBJ accession numbers for the genome sequence is JABULH000000000. The DNA G + C content is 67.1%.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Asaf S, Numan M, Khan AL, Al-Harrasi A (2020) Sphingomonas: from diversity and genomics to functional role in environmental remediation and plant growth. Crit Rev Biotechnol 40:138–152
Aylward FO, McDonald BR, Adams SM et al (2013) Comparison of 26 sphingomonad genomes reveals diverse environmental adaptations and biodegradative capabilities. Appl Environm Microbiol 79:3724–3733
Collins MD, Jones D (1980) Lipids in the classification and identification of coryneform bacteria containing peptidoglycans based on 2, 4-diaminobutyric acid. J Appl Microbiol 48:459–470
Collins MD, Pirouz T, Goodfellow M, Minnikin DE (1977) Distribution of menaquinones in actinomycetes and corynebacteria. J Gen Microbiol 100:221–230
Cowan ST, Steel KJ (1996) Manual for the identification of medical bacteria. Cambridge University Press, London, p 232
Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17:368–376
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Feng GD, Yang SZ, Zhu HH, Li HP (2018) Emended descriptions of the species Sphingomonas adhaesiva Yabuuchi et al. 1990 and Sphingomonas ginsenosidimutans Choi et al. 2011. Int J Syst Evol Microbiol 68:970–973
Fitch WM (1971) Toward defining the course of evolution: minimum change for a specific tree topology. Syst Biol 20:406–416
Galperin MY, Makarova KS, Wolf YI, Koonin EV (2015) Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res 43:D261–269
Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM (2007) DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57:81–91
Huang HY, Li J, Zhao GZ et al (2012) Sphingomonas endophytica sp. nov., isolated from Artemisia annua L. Int J Syst Evol Microbiol 62:1576–1580
Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, von Mering C, Bork P (2017) Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol Biol Evol 34:2115–2122
Khan AL, Waqas M, Kang S-M et al (2014) Bacterial endophyte Sphingomonas sp. LK11 produces gibberellins and IAA and promotes tomato plant growth. J Microbiol 52:689–695
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549
Lane DJ (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. Wiley, Chichester, pp 115–175
Larkin MA, Blackshields G, Brown NP et al (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Li YQ, Rao MPN, Zhang H et al (2019) Description of Sphingomonas mesophila sp. nov., isolated from Gastrodia elata Blume. Int J Syst Evol Microbiol 69:1030–1034
Meier-Kolthoff JP, Auch AF, Klenk HP, Goker M (2013) Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform 14:60
Minnikin DE, Collins MD, Goodfellow M (1979) Fatty acid and polar lipid composition in the classification of Cellulomonas, Oerskovia and related taxa. J Appl Microbiol 47:87–95
Nurk S, Bankevich A, Antipov D et al (2013) Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol 20:714–737
Okonechnikov K, Golosova O, Fursov M, Team U (2012) Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28:1166–1167
Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil PA, Hugenholtz P (2018) A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol 36:996–1004
Parte AC (2018) LPSN - List of Prokaryotic names with Standing in Nomenclature (bacterio.net), 20 years on. Int J Syst Evol Microbiol 68:1825–1829
Price MN, Dehal PS, Arkin AP (2009) FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26:1641–1650
Richter M, Rossello-Mora R (2009) Shifting the genomic gold standard for the prokaryotic species definition. P Natl Acad Sci USA 106:19126–19131
Rivas R, Abril A, Trujillo ME, Velazquez E (2004) Sphingomonas phyllosphaerae sp. nov., from the phyllosphere of Acacia caven in Argentina. Int J Syst Evol Microbiol 54:2147–2150
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sasser M (1990) Identification of bacteria by gas chromatography of cellular fatty acids, MIDI Technical Note 101. Microbial ID, Inc, Newark
Siddaramappa S, Viswanathan V, Thiyagarajan S, Narjala A (2018) Genomewide characterisation of the genetic diversity of carotenogenesis in bacteria of the order Sphingomonadales. Microb Genom 4:e000172
Sievers F, Wilm A, Dineen D et al (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539
Tamaoka J (1986) Analysis of bacterial menaquinone mixtures by reverse-phase high-performance liquid chromatography. Methods Enzymol 123:251–256
Tatusova T, DiCuccio M, Badretdin A et al (2016) NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 44:6614–6624
The Gene Ontology Consortium (2019) The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res 47:D330–D338
Yang S, Zhang X, Cao Z et al (2014) Growth-promoting Sphingomonas paucimobilis ZJSH1 associated with Dendrobium officinale through phytohormone production and nitrogen fixation. Microbial Biotechnol 7:611–620
Yoon SH, Ha SM, Kwon S, Lim J, Kim Y, Seo H, Chun J (2017) Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol 67:1613–1617
Acknowledgements
This research was supported by the National Natural Science Foundation of China (Nos. 31900001 and 31972856). D-F Zhang was also supported by the Fundamental Research Funds for the Central Universities (2019B02014)
Author information
Authors and Affiliations
Contributions
DFZ and ZZ designed research and project outline. XWC, DFZ, and AHZ performed isolation, deposition and polyphasic taxonomy. DFZ, and JH performed genome analysis. DFZ and AHZ drafted the manuscript. WJL revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, DF., Cui, XW., Zhao, Z. et al. Sphingomonas hominis sp. nov., isolated from hair of a 21-year-old girl. Antonie van Leeuwenhoek 113, 1523–1530 (2020). https://doi.org/10.1007/s10482-020-01460-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-020-01460-z